Experimental Animal Study
All experimental procedures were conducted at the Stem Cell Research Center affiliated with Yıldırım Beyazıt Training and Research Hospital (Ankara, Türkiye). The study protocol was approved by the Ethics Committee of Yıldırım Beyazıt Training and Research Hospital and was carried out in accordance with the Guide for the Care and Use of Laboratory Animals and the European Parliament Directive 2010/63/EU.
Fifty female Sprague-Dawley rats, eight weeks old and weighing between 150 and 180 g, were used in the experiment. The animals were randomly assigned to five experimental groups (n = 8 per group). In addition, 10 female rats of the same age and weight range were allocated specifically for the isolation of mesenchymal stem cells from fetal tissue (gestational day 16). The experimental groups were as follows:
1.
Control Group (n = 8): Healthy rats without any intervention.
2.
Diabetes Group (n = 8): Rats induced with diabetes via a single intraperitoneal injection of streptozotocin (STZ, 45 mg/kg).
3.
OMSC Treatment Group (n = 8): Diabetic rats treated with ovary-derived mesenchymal stem cells.
4.
EMSC Treatment Group (n = 8): Diabetic rats treated with endometrium-derived mesenchymal stem cells.
5.
BMMSC Treatment Group (n = 8): Diabetic rats treated with bone marrow-derived mesenchymal stem cells.
The rats were housed under controlled environmental conditions at a temperature of 20–24 °C, with 40–60% relative humidity, and maintained on a 12-hour light (06:00–18:00) and 12-hour dark cycle. All animals were fed a standard rat chow diet and had ad libitum access to water. They were kept in group-housed cages with clean bedding material, and every effort was made to minimize discomfort and pain throughout the study.
Induction of Experimental Diabetes Model
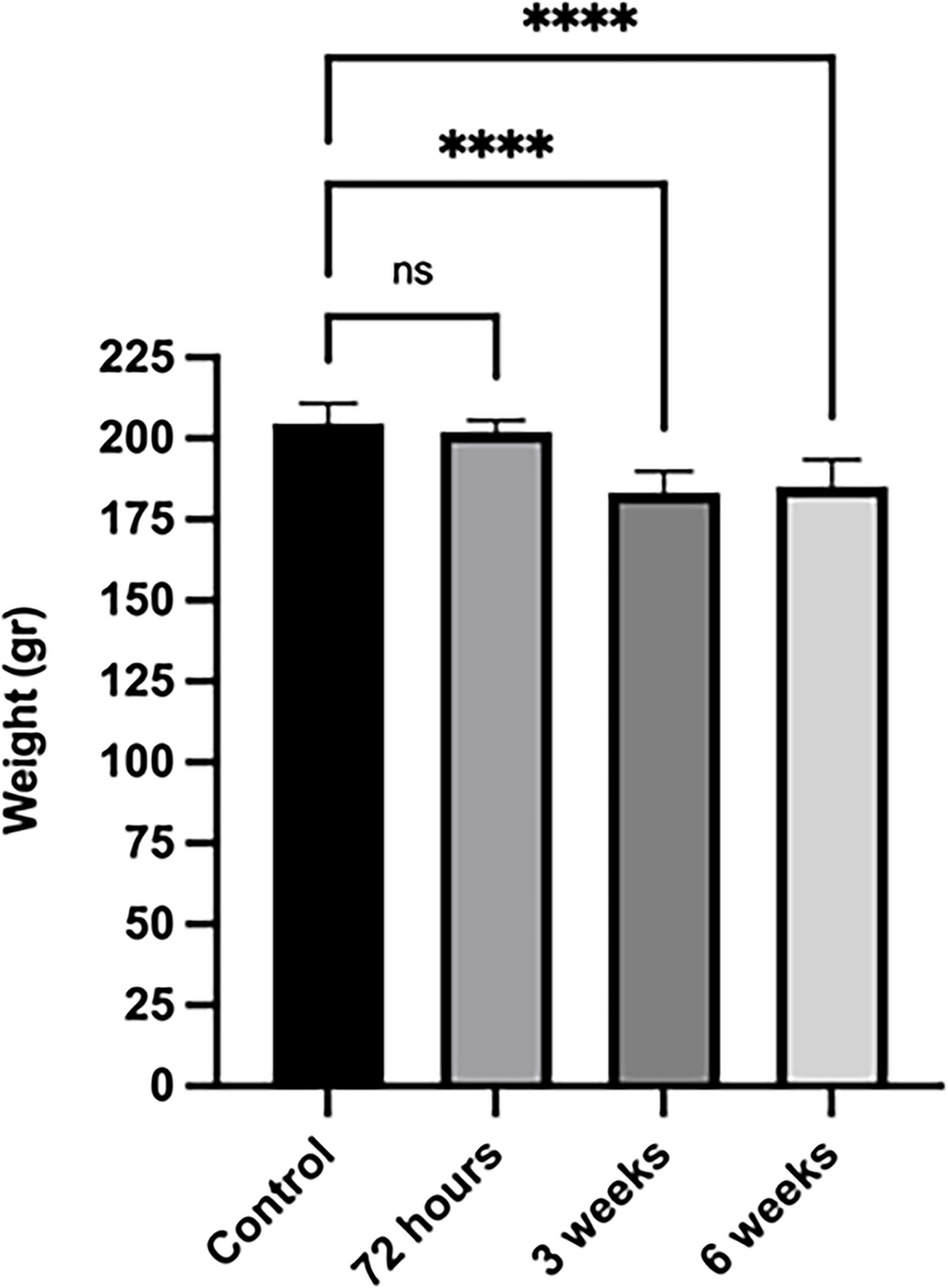
To induce diabetes, 32 female Sprague-Dawley rats were administered STZ (Sigma-Aldrich) at a dose of 45 mg/kg, dissolved in 0.1 mmoL sodium-citrate buffer (pH 4.5), via intraperitoneal injection. To confirm the development of diabetes, blood glucose levels were measured from tail vein blood samples 72 h after injection, and rats with blood glucose levels of 200–250 mg/dL or higher were considered diabetic. The diabetic rats were monitored for 6 weeks, with blood glucose levels and body weights measured and recorded at the 72nd hour, 3rd week, and 6th week, both in the morning and evening [22].
Mesenchymal Stem Cell Isolation
Following deep ketamine/xylazine anesthesia, the abdominal area of the Sprague-Dawley rats (gestational day 16) was cleaned with Betadine® (10% povidone-iodine solution; Mundipharma), an antiseptic commonly used for skin disinfection prior to surgical procedures, and neonatal ovarian and endometrial tissues were collected. Immediately after collection, the tissues were placed into a transport medium and transferred without delay to the laboratory. Upon arrival, the tissues were placed in a glass Petri dish and cut into small pieces using a scalpel. They were then digested into a single-cell suspension using 1 mg/mL collagenase type I (Sigma, Missouri, United States) and transferred to the bottom of a T25 flask. After incubating for 15–30 min, the tissue digests were sequentially filtered twice through 100 μm cell strainers (Corning, New York, United States) and then through a 40 μm cell strainer (Corning), effectively removing undigested tissue and epithelial cells at each step. Finally, mesenchymal stem cell culture medium was gently added. The culture medium was changed every three days, and passaging was performed based on cell growth and development [23–24].
Under deep ketamine/xylazine anesthesia, the 16-day-old fetal Sprague-Dawley rats designated for mesenchymal stem cell isolation were euthanized. The abdominal region was disinfected with Betadine® (10% povidone-iodine solution), and neonatal bone marrow material was collected from the femurs and tibias into a transport medium immediately after extraction. Both ends of each bone were fractured under sterile conditions, and the marrow cavity was flushed using 5 mL syringes filled with mesenchymal stem cell culture medium. The bone marrow suspension was collected into sterile Falcon tubes. Ficoll was added to new Falcon tubes, and the collected bone marrow was slowly layered onto the Ficoll solution. The samples were centrifuged at 1200 rpm for 5 min. The resulting buffy coat was collected into new tubes, and the volume was adjusted to 10 mL with mesenchymal stem cell culture medium. The samples were centrifuged twice at 1200 rpm for 5 min each time. The supernatant was discarded, and fresh mesenchymal stem cell culture medium was added to the cell pellet. The cells were seeded into T25 flasks. The culture medium was changed every three days, and passages were performed based on cell growth and confluence [24–25].
The stem cells isolated during the experimental process were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% (v/v) heat-inactivated fetal bovine serum (FBS), 20 mM L-glutamine, and 1% penicillin-streptomycin.
Characterization of Mesenchymal Stem Cells Using Flow Cytometry
In this study, the flow cytometry method was utilized to determine the differentiation potential of primary cells into multipotent cell types such as osteoblasts, chondrocytes, and adipocytes. For this purpose, cells were suspended after the second passage to a concentration of 1 × 10⁶ cells in 100 µL aliquots, and 10 µL of surface markers (CD45, CD11b/c, CD44, CD90; all markers from BD-USA) were added and vortexed. The samples were incubated in the dark at 4 °C for 15 min. Following incubation, 2 mL of washing solution was added, and the samples were centrifuged at 1200 rpm for 10 min, after which the supernatant was discarded. To the cell pellet, an additional 1 mL of washing solution was added, and the samples were characterized using a flow cytometry device (FACS ARIA III, Becton, Dickinson and Company, Vancouver, Canada) [24, 26].
Characterization of Mesenchymal Stem Cells by Tri-lineage Differentiation Assay
To confirm differentiation into adipocyte cells, the cells were cultured for three weeks using Adipocyte Differentiation Basal Medium and supplements (Gibco, USA), and lipid droplets were visualized in red using the Oil Red staining method (Diagnostic BioSystem, USA). To determine differentiation into chondrocyte cells, Chondrocyte Differentiation Basal Medium was utilized, and the cells were cultured for three weeks, with the presence of hyaluronic acid detected by staining with Alcian Blue (Diagnostic BioSystem, USA), resulting in a blue coloration. The differentiation of osteocyte cells was assessed by culturing them with Osteocyte Differentiation Basal Medium, followed by the Von Kossa staining method (Diagnostic BioSystem, USA), which stained calcium deposits black [27,28,29].
Application of Stem Cells in Diabetic Rats
To track the localization of stem cells administered to diabetic rats at the end of the experiment and to evaluate their contribution to healing, mesenchymal stem cells were stained with BrdU (Bromodeoxyuridine). For the staining procedure, a 10 µM BrdU solution was prepared at a concentration of 1 mL. The cells were incubated for 2 h (37 °C, 5% CO₂) by adding 10 µL of the BrdU solution to each 1 mL containing 2 × 10⁶ cells. The identified mesenchymal stem cells were labeled with BrdU and injected into diabetic rats induced with STZ at the end of the third week via the endometrial canal to the ovary, at a concentration of 2 × 10⁶ cells/mL PBS in a total volume of 1 mL. This procedure was repeated in the fourth week. At the end of the sixth week, the abdominal cavities of the rats were surgically opened, the ovarian tissues were excised, and fixation was performed using 4% formaldehyde. For histological and immunohistochemical analyses, all rat groups were anesthetized via intraperitoneal injection of ketamine and xylazine. The abdominal cavities of the rats were opened, and the ovarian tissues were excised and fixed in 4% formaldehyde. The fixed tissues were dehydrated using increasing concentrations of ethanol. Subsequently, the tissue was embedded in melted paraffin at 60 °C, and blocks were prepared. The prepared blocks were sliced into 4 μm thick sections using a microtome [24, 30].
Histochemical Analyses Using Hematoxylin & Eosin (H&E) Staining
Sections with a thickness of 4 μm obtained from paraffin blocks were subjected to routine histological procedures. For the deparaffinization stage, the sections were incubated overnight at 37 °C, followed by an additional hour at 57 °C. After this process, the sections were placed in xylene three times for 20 min each and then rehydrated in a series of decreasing ethanol concentrations (100%, 90%, 80%, 70%, and 50%), each for 10 min. Following air drying, the sections were washed with tap water for 10 min to remove any residual ethanol. Subsequently, the sections were incubated in Harris hematoxylin staining solution for 10 min, rinsed again under tap water for 10 min, and immersed 2–3 times in a mixture of glacial acetic acid and ethanol before being washed under tap water for another 10 min. Following this, the sections were treated in Eosin staining solution for 10 min and then washed again with tap water for 10 min. The dehydration process was completed by quickly passing the sections through increasing concentrations of alcohol (50%, 70%, 80%, 90%, and 100%). Finally, the sections were treated with xylene for 15 min and mounted with Entellan. The images obtained from the prepared sections were captured using a Leica DM 4000B (Germany) computer-assisted light microscope and evaluated using the Leica LAS V4.9 software [31].
Immunohistochemical Analyses
The immunohistochemical method was performed using the Ultra Vision Detection System Large Volume Anti-Polyvalent, HRP (RTU) and the Ultra Vision Detection System DAB Substrate kits. For immunohistochemical staining, 4 μm thick sections were taken from paraffin blocks and placed on polylysine-coated slides. After the deparaffinization stage, the sections were processed through a series of decreasing alcohol concentrations of 100%, 96%, 90%, 80%, and 70%. The dehydrated tissues were rinsed twice for 5 min with distilled water to remove the ethanol, followed by antigen retrieval using citrate buffer (pH 6.0). To remove the citrate buffer, the tissues were washed twice again with distilled water for 5 min each. The sections were then placed in an immunohistochemistry chamber surrounded by a PAP pen to maintain a moist environment. The tissues were washed three times for 3 min each with phosphate-buffered saline (PBS, pH 7.4), and then treated with 3% hydrogen peroxide for 15 min. After this treatment, the slides were washed with PBS and incubated with UltraV block (TA-125-UB, Lab Vision, Fremont, CA) for 5 min to prevent nonspecific binding. Without washing, the primary antibody application was performed, and the sections were incubated overnight with VEGF primary antibody (bs-1665R, Bioss, USA) at an appropriate dilution (1:200). Additionally, for stem cell labeling, the sections were incubated for 60 min at room temperature with bromodeoxyuridine (BrdU) primary antibody (550803; BD Pharmingen) at a dilution of 1:10. After the primary antibody application, the slides were washed three times with PBS for 3 min each. Following the wash, a biotinylated secondary antibody (TP-125-BN, Lab Vision, Fremont, CA) was applied for 10 min, followed by treatment with the streptavidin-peroxidase enzyme complex (TS-125-HR, Lab Vision, Fremont, CA) for 10 min. Finally, the chromogen diaminobenzidine (DAB) (TA-125-HD, Lab Vision, Fremont, CA) was applied to visualize the immunoreaction. Mayer’s hematoxylin was used as a counterstain, and the slides stained with DAB were processed through decreasing alcohol series. After being kept in xylene for 20 min, the slides were mounted with Entellan and analyzed using the Leica DM 4000 (Germany) computer-assisted imaging system and the Leica Q LAS software [32].
Follicle Counting and Immunopositivity Determination Method
For follicle counting and immunohistochemical assessment, ovarian tissues were sectioned serially into 4 μm thick slices. Every fifth slice was used for follicle counting, while the sixth slice was utilized for immunohistochemical evaluation. In all ovarian sections stained with Hematoxylin and Eosin (H&E), the counts of primordial, primary, and antral follicles were recorded. As part of the immunohistochemical assessment, the percentage (%) of VEGF immunostaining in each section was calculated using Image J software [33].
Statistical Analyses
Data were entered into the SPSS 21.0 statistical software, and multiple ROC curve analyses were performed. A p-value of less than 0.05 was considered statistically significant.


Comments (0)