
Remember me


In total, 21 urine DNAs were whole-genome sequenced with native methylation calling using the ONT platform. Initially, 9 BCs and 5 non-BCs were sequenced at one sample per flow cell, providing median genome coverages of 18-34x. These data suggested that lower read depths might be sufficient for BC detection and a further 8 samples (4 BCs and 4 non-BCs, including a non-BC repeat sample) were multiplexed at 4 samples per flow cell, providing median genome coverages of 2-5x. The mean read length was 1.8 kb (range 0.7 kb to 3.9 kb) (Supplementary Table S1). A non-BC sample was sequenced twice (at both high and low coverage) to investigate the effect of read depth on CpG modification fraction and copy number events. The Pearson correlation coefficient between methylation fraction across 42.9 M CpG positions sequenced in both the high and low coverage for this sample was 0.558 (p-value < 2.2 × 10–16) (Supplementary Fig. S1). When considering the 10,479 DMRs, the correlation coefficient increased to 0.717. Correlation coefficients were also > 0.6 with all non-BC samples and negative correlations were seen with all high VAF BC samples.
Differential urine DNA methylation in BC and non-BC patientsWe identified 0.73 million genomic segments with differential CpG methylation between the BC and non-cancer patients. Further filtering on DMR length, HMM score, modified CpG positions count and modification fraction resulted in 0.2 million segments. Mann–Whitney tests were used to identify a high-confidence set of 10,479 DMRs of which 4048 were hypermethylated and 6431 hypomethylated in BC. To confirm our DMR search strategy, we plotted separately the hyper- and hypo-DMR segments with their respective genomic flanking regions (± 500 bp), observing the expected hyper- or hypomethylated profiles (Supplementary Fig. S2A-B). Differential methylation has been reported to appear frequently at transcription start sites, reflecting differential regulation of gene expression between normal and cancer cells [20]; indeed, this was captured by our DMR approach (Supplementary Fig. S2C).
We used Mann–Whitney tests to triage DMRs; however, these did not retain statistical significance after Benjamini–Hochberg multiple testing correction, most likely due to the modest sample size, extremely high number of possible DMRs, disease molecular heterogeneity, different stages and grades of disease included, and the variable tumour DNA fraction in patient samples. In order to determine if our observations as a whole do indeed capture biologically relevant signals, we compared the number of detected DMRs to a random background model (50-fold sample randomisation), as well as the number of enriched pathways within these sets (50-fold DMR random selection). The median number of DMRs from these random group comparisons was 140, strongly suggesting that the majority of our 10,479 DMRs are not chance findings (Supplementary Fig. S3A). The median number of significant pathways observed was 0 for the random segment sets, compared to 8 in the actual analysis (Supplementary Fig. S3B).
Global hypomethylation and promoter hypermethylation in BCThe majority of the DMR segments were hypomethylated (61%) in BC, covering 14 Mb of the genome, whereas hypermethylated segments (39%) covered 5 Mb. The average size of hypermethylated segments was 1.2 kb versus 2.2 kb for hypomethylated segments. Hypermethylated segments showed greater absolute changes in methylation levels than hypomethylated segments: average delta values of 2 vs. 0.7, respectively (Mann–Whitney p-value < 2.2e-16).
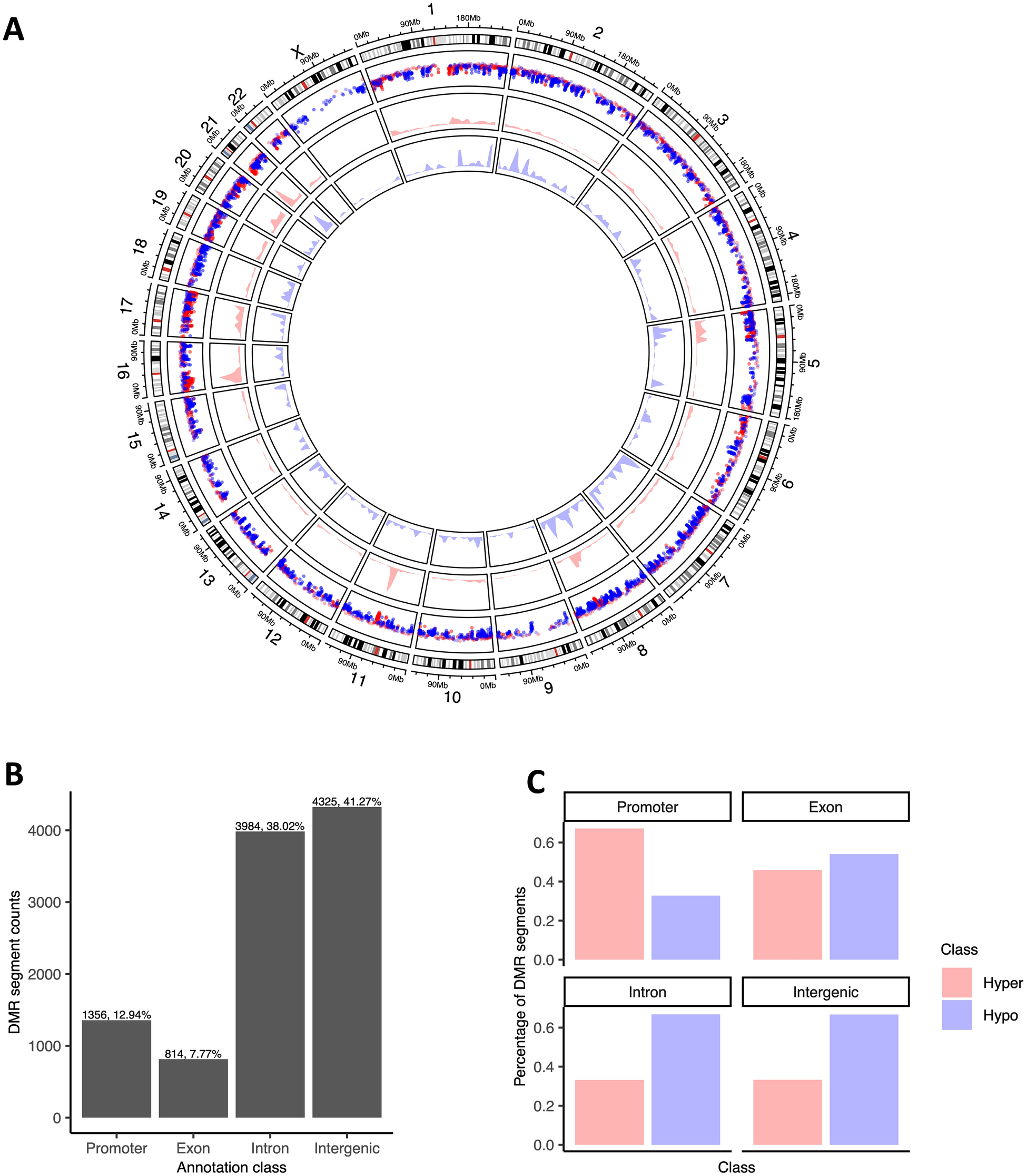
DMRs were observed across all chromosomes (Fig. 1A), but their distribution was not directly related to chromosome size or gene content, indicating non-randomness of the methylation events: the chromosomes with the most hypermethylated DMRs were chr1, chr17, chr5 and chr6 (10, 9, 6.5 and 6.5% of all hypermethylated DMRs, respectively), and the chromosomes with the most hypomethylated DMR content were chr2, chr7 and chr8 (9.8, 9.6 and 9%, respectively). Annotation of DMRs for genomic context revealed that 13% were in promoter regions (± 3 kb of TSS for 1036 unique genes), 8% exonic (for 589 unique genes), 38% intronic (for 1677 unique genes) and 41% intergenic (covering 7.9 Mb of intergenic space) (Fig. 1B). DMRs were frequently hypermethylated in gene promoters, whereas in other regions hypomethylation dominated (Fig. 1C); hypermethylated DMRs (versus hypomethylated DMRs) were around 4 times more likely to be in promoter regions, whereas hypomethylated DMR segments were 2 × more likely to be in intergenic regions (Chi-square test p-value < 2.2e-16; Supplementary Fig. S4).
Fig. 1
A Circos plot of genome-wide methylation and copy number changes in BC patient urinary DNA. The outermost track is the chromosome ideogram. Moving from the outer to the circle centre, the 2nd track is a rainfall plot of DMRs across the chromosomes (red and blue indicate hyper- and hypomethylation, respectively). The 3rd and 4th tracks are the density plots for hyper- and hypomethylated DMRs, respectively. B Distribution of DMR counts across the genomic context classes. C Comparative distribution of hypermethylated versus hypomethylated segments per genomic context annotation class
DMRs in urine DNA distinguish bladder cancer patients from non-cancer patientsPrincipal component analysis of the DMRs separated BC and non-cancer urine samples (Fig. 2A). Five BC samples with high-coverage and high tumour content (VAFs > 30%) clearly separate from the high-coverage non-BC samples in PC1. Four NMIBC samples with low coverage and VAFs of 13–25% segregate with the high VAF high-coverage NMIBC samples, demonstrating that DMR segments can provide cancer-specific signals even in samples with low sequencing depth. Similarly, the low-coverage non-cancer samples segregate with the high-coverage non-cancer samples. Four high-coverage NMIBC samples with lower VAFs lie between the non-BC and higher VAF samples. We also analysed the data by h-clust obtaining a BC cluster and a predominantly non-BC cluster (Fig. 2B). The BCs which cluster with the non-BCs are the same low VAF high-coverage BCs which are close to the non-BCs in the PCA. Commonly reported hypermethylation markers [21] behave as expected in our data (Fig. 3).
Fig. 2
Principal component analysis (PCA) and hierarchical clustering of BC and non-BC urine DNA methylation. A PCA plot with the X- and Y-axes representing PC1 and PC2, respectively. Non-BCs are represented by green and BCs by pink symbols. The shapes of the data points indicate whether the samples have low or high sequencing depth and indicate low or high variant allele frequency cases for BC samples. B Heatmap showing hierarchical clustering of DMRs. X- and Y-axes represent the samples and DMR segments, respectively
Fig. 3
Nanopore methylation data across known BC biomarkers. The boxplot shows the methylation status (determined by ONT direct calling) for 5 gene promoters reported as hypermethylated in BC in a recent systematic review. The 5mC modification fraction (aggregate signal value for all constituent CpG positions in log2 scale; Y-axis) in the 1 kb upstream and downstream of TSS (transcription start site) was plotted separately for non-BC and BC samples, for the five different biomarkers (X-axis). The dots represent the non-BC (n = 8) or BC (n = 13) samples in the cohort
All BC urine samples were positive for ≥ 1 SNV when previously deep-sequenced with GALEAS Bladder. Although the ONT sequencing read depth is insufficient for sensitive SNV detection, 21 of the 31 SNVs detected in the BC urines by GALEAS Bladder (68%) were also supported by ≥ 1 read in the 9 high-coverage BC urine samples, and 12 out of 29 (41%) in the low-coverage BC urine samples.
Gene set enrichment analysis (GSEA) of DMRsThe DMR segments included the promoter and/or gene body regions of 2,769 genes—we ranked these genes by methylation delta value to reflect the difference between BC and non-BC methylation. GSEA using the ranked methylation data on the 50 Hallmark (MSigDb) pathways revealed positive enrichment of 8 pathways (adjusted p-value < 0.05) for hypermethylation signal (Fig. 4 and Supplementary Table S2). The most significant pathways were mitotic spindle, G2M checkpoint and hypoxia. These data suggest that genome-wide urine DNA methylation patterns could be used for the non-invasive molecular characterisation of primary bladder tumours.
Fig. 4
Gene set enrichment analysis (GSEA) of DMRs using MSigDb Hallmark pathways. Of the 50 pathway sets, 8 were found to be significant at threshold of adjusted p-value < 0.05 and normalised enrichment score (NES) > = 1.5 or < = − 1.5. The above plot shows all the pathways crossing the NES threshold irrespective of adjusted p-value. The X- and Y-axes denote NES and the pathway names, respectively. The size of the given dot for a given pathway is indicative of the number of genes (legend “size”) observed from the pathway, and the colour is indicative of the adjusted p-value (legend “padj”)
Long-read sequencing provides comprehensive coverage of methylation in repetitive elementsIn each sample, we observed a uniform read depth regardless of the repetitive sequence content (average difference between repetitive and non-repetitive: 0.028x, with standard deviation of 0.036), demonstrating that long-read sequencing enables unbiased investigation of these regions with reliable coverage after alignment. This ability to circumvent potential blind spots caused by repetitive elements—an issue potentially encountered in short-read sequencing due to mapping difficulties [11]—proved advantageous in our analysis. Notably, approximately 59% of the sequence within intergenic region DMRs, which tend to be hypomethylated in BC, consisted of repetitive elements, compared to 27% in exonic DMRs and 30% in promoter DMRs. (Supplementary Fig. 5A). Furthermore, the repetitive element content of hypomethylated DMRs was significantly higher than the hypermethylated DMRs (Mann–Whitney p-value < 2.2e-16) (Supplementary Fig. 5B) and the difference in distribution of the repetitive element content in the two DMR classes was more prominent in promoter and exonic regions (Supplementary Fig. 5C).
Copy number variants in BC patient ucpDNASince ucpDNA is extracted from an admixture of healthy and tumour cells, we hypothesised that copy number detection methods can be effectively applied, provided the tumour content is not too low. High VAF BC urine samples were easily distinguishable from non-cancer patients in genome-wide karyotype profile plots, but less so for low VAF cases (Fig. 5 and Supplementary Fig. 6A-D); the BC urine genomes harboured more CNVs than the non-BCs (20.8% vs. 0.04% of the genome altered; Mann–Whitney p-value 0.001) (Supplementary Fig. 6E). Amplification of chr8q was observed in 61% of BCs, and across these samples, the chr8q22.2 region was consistently altered. Chr9p aneuploidy was observed in 46% of samples. On chr5, the p arm tended to show gains (38%), while the q arm tended to be lost (38%). Chr17 showed a similar pattern with losses frequently seen on the p arm (38%) and typically gains on the q arm (Fig. 1A). The changes on chr9, chr5 and chr17 were not detected in any low VAF BC samples.
Fig. 5
Copy number changes in DNA from the urine of BC patients and control subjects. The figure shows copy number event calls made in non-bladder cancer (non-BC) and bladder cancer (BC) samples. The Y-axis has the chromosome locations (chr1-22 in the positive direction), with samples arrayed along the X-axis
Comments (0)