
Remember me
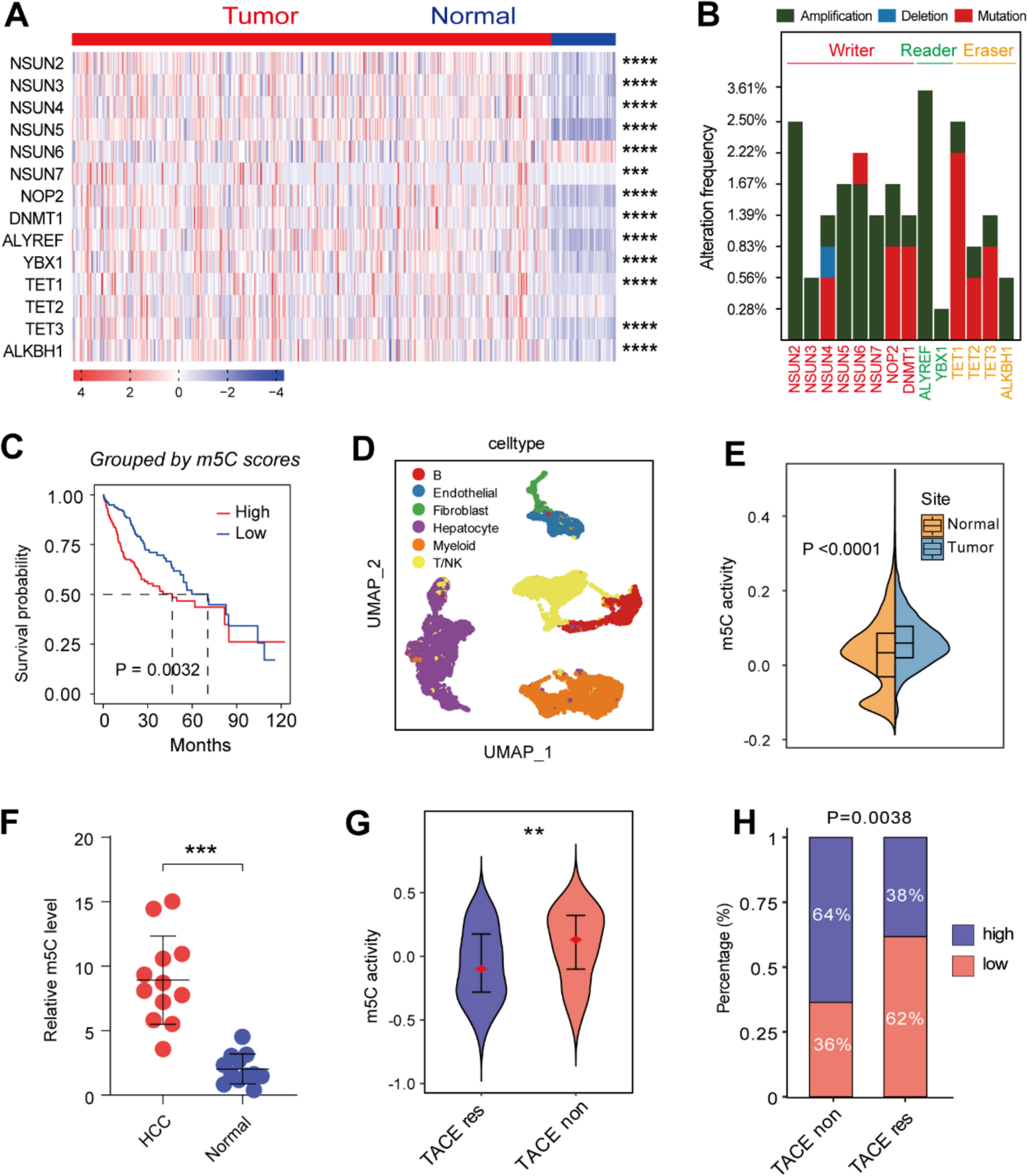


Fourteen m5C regulators were identified from the results of previous studies, including the m5C methyltransferases/writers NSUN2-7, NOP2, and DNMT1, the binding proteins/readers ALYREF and YBX1, and the demethylases/erasers TET1-3 and ALKBH1. The mRNA expression levels of these m5C regulators were then compared using transcriptomic data from the The Cancer Genome Atlas-Liver Hepatocellular Carcinoma (TCGA-LIHC) cohort. This showed marked differential expression of most of the regulators between HCC and normal tissues (Fig. 1A). The patterns of change in m5C regulator expression in HCC tissues were then analyzed using the online tool cBioPortal (Fig. 1B). Overall, the m5C regulators showed relatively low frequencies of altered expression, with changes falling into three categories, namely, amplification, deletion, and mutation. To explore the potential impact of m5C modifications on clinical prognosis, an analysis of overall survival (OS) in relation to the m5C regulators was performed using TCGA data (Supplementary Figure S1A–C). Kaplan–Meier survival curves showed significant associations between many of the m5C regulators and survival in patients with HCC. K–M curve based on overall m5C scores validated that patients with higher scores have shorter OS, respectively in TCGA and GSE54236 dataset (Fig. 1C; Supplementary Figure S1D). Moreover, single cell RNA sequencing (scRNA-seq) data from GSE149614 was analyzed to visualize overall m5C levels in tumor and normal sites (Fig. 1D; Supplementary Figure S1E), which manifested significantly higher levels of m5C in tumor tissues (Fig. 1E; Supplementary Figure S1F). We subsequently analyzed the correlation of m5C regulators and hallmark pathways, which revealed the potential participation of m5C modification in multiple significant signalling pathways (Supplementary Figure S2A). Samples from HCC patients validated our finding (Fig. 1F). These results confirmed that m5C modification was abnormally elevated in HCC and associated with worse prognosis.
Fig. 1
Comprehensive profiling of the significance of m5C modification in HCC. A Heatmap showing a comparison of m5C regulator mRNA expression levels between HCC and normal tissues, based on TCGA data. B Frequencies of genetic alterations in m5C regulators. C K–M curve shows significant difference between m5Chigh and m5Clow groups. The threshold score for dividing high and low groups was − 0.559, which was the median score. D UMAP plot of scRNA-seq data from GSE149614. Total cell counts are 16,244. E Comparison of m5C scores between tumor and normal sites in scRNA-seq data. F Comparison of m5C levels in twelve pairs of HCC and normal samples. G Comparison of m5C scores between TACE responders and non-responders. H Stack plot shows proportion of m5Chigh versus m5Clow in responder and non-responder groups. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001)
Next, to further investigate the roles of m5C modification in HCC, we utilized GSE104580 dataset to explore whether m5C modifications affect clinical treatment. We found that transcatheter arterial chemoembolization (TACE) treatment responders have overall lower m5C scores compared with non-responders (Fig. 1G, H). Moreover, the AUC area was 0.654, which represented well efficiency of m5C score for TACE response prediction. Together, our findings revealed the significant influence of m5C modification on HCC prognosis and clinical treatment.
Identification of three distinctive m5C modification clusters and their respective characteristicsUnsupervised clustering was then performed to analyze m5C modification patterns in HCC tissues using the “ConsensusClusterPlus” package in R. This showed that all samples were clustered into three clusters, represented as cluster 1 (n = 61), cluster 2 (n = 159), and cluster 3 (n = 154) (Fig. 2A–C). Principal component analysis (PCA) was then performed to verify the findings (Supplementary Figure S2A). The three m5C clusters were then analyzed using Kaplan–Meier curves. Consistent with the previous findings, cluster 1, which had the highest expression of most m5C regulators, was associated with the lowest OS rates, followed by cluster 2 (Fig. 2D). Also, the expression levels of m5C regulators were compared among the different clusters. This showed that the levels of most regulators were significantly upregulated in cluster 1, with intermediate levels in cluster 2, and the lowest levels in cluster 3 (Fig. 2E), suggesting that cluster 1 had the highest levels of m5C modification, followed by cluster 2, and then cluster 3. To examine differences in immune functions among the different clusters, the levels of immune cell infiltration (Supplementary Figure S1B)., stromal scores, immune scores, and ESTIMATE scores were evaluated (Supplementary Figure S1C–E). It was found that higher levels of m5C modification, were associated with greater infiltration of antitumor immune cells (Supplementary Figure S1B). However, the stromal, immune, and ESTIMATE scores did not differ significantly among the clusters. Genomic alterations associated with the three clusters were also analyzed in Supplementary Figure S2F which indicated that patients with different m5C modification activity might have different patterns of alterations. Evidence shows that m5C modifications influence cell metabolism and signal transduction [27, 28]. Also, differences in OS among the clusters were not specifically associated with immune differences, we thus assessed the Gene Set Variation Analysis (GSVA) scores of multiple metabolism pathways and classic pathways associated with oncogenic activities (Fig. 2F; Supplementary Figure S4A, B). As a result, all clusters exhibited completely different characteristics in metabolic perspective, and higher m5C activity aligned with higher oncogenic activities. In this part, our findings confirmed 3 clusters associated with m5C modifications, and comprehensively described their immune, genomic alterations and metabolic characteristics. These findings suggested that m5C modification could potentially activate oncogenic and metabolism pathways.
Fig. 2
Identification of m5C modification-associated clusters by unsupervised consensus clustering. A Cumulative distribution function (CDF) displays the stability of clustering across k = 2–7. B Delta area plot displays the area under the CDF curve for different k-values, with k = 3 identified as the optimal k-value. C Consensus matrix (k = 3) heatmap demonstrating clustering consistency, with distinct borders indicating distinct and well-defined clustering. D Kaplan–Meier curves indicate distinct differences in OS among the m5C modification clusters. E Comparisons of m5C regulator expression levels in the three m5C clusters. P-values were determined by Kruskal–Wallis tests. F Heatmap showing the differences of multiple signaling pathways and dysregulated metabolism pathways across clusters. Data were analyzed by Kruskal–Wallis tests. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001)
Metabolic landscape profiles of the m5C modification clusters show DNMT1-mediated dysregulated pyrimidine metabolismOur previous work evaluated the GSVA scores of traditional pathways and metabolic characteristics of the three m5C clusters (Fig. 2F; Supplementary Figure S4A). This indicated significant activation of most traditional pathways in the highest m5C cluster (C1), together with relative suppression in the lowest cluster (C3) (Fig. 2F). Notably, it was observed that pyrimidine metabolism pathways (Fig. 3A) showed significantly different distribution patterns in the three m5C clusters, highest in cluster 1, intermediate in cluster 2, and lowest in cluster 3 (Fig. 3B). It was thus hypothesized that abnormally raised levels of m5C modification stimulate pyrimidine metabolism. To test this hypothesis, the expression of key enzymes associated with pyrimidine metabolism (Fig. 3C) was compared between the three clusters. As shown in the heatmap, the expression of these enzymes showed a positive correlation with m5C levels, aligning with previous findings.
Fig. 3
Comprehensive profiling of the metabolic characteristics of the m5C clusters showed that DNMT1 mediated dysregulated pyrimidine metabolism. A The process of pyrimidine biosynthesis, from materials to DNA synthesis. Key enzymes are shown in red. B Comparisons of GSVA scores of pyrimidine metabolism. C Z-scores of mean expressions of key enzymes of pyrimidine biosynthesis. D Ranks of fold change of m5C regulators in cluster1. E Differences in m5C regulator expression levels in clinical samples of HCC and normal tissues. DNMT1 exhibited most distinct difference. F Relative expression of DNMT1 in six HCC cell lines. G, H DNMT1 expression levels after DNMT1 knockdown in MHCC97H and Huh7 cells, shown by RT-PCR (E) and western blotting (F). I Comparison of relative m5C levels between the control and DNMT1-knockdown groups. J, K Comparison of key pyrimidine metabolism-associated regulators expression between the control and DNMT1-knockdown groups, shown by RT-PCR (J) and WB (K). L Heatmap showing changes in metabolites of pyrimidine metabolism between control and DNMT1 knockdown group. M, N Colony formation assay and transwell assay results showing colony formation (K) in Huh7 and MHCC97H cells, as well as migration and invasion in MHCC97H cells (L) after DNMT1 knockdown. (*P < 0.05, ** P < 0.01, ***P < 0.001)
Next, we aimed to investigate whether an m5C methyltransferases changed the status of m5C modifications in cluster 1 to trigger the upregulated pyrimidine metabolism. We first conducted differentially expressed genes (DEG) analysis, which showed that DNMT1 was most elevated (Fig. 3D). The expression of m5C regulators in normal and HCC tissues reinforced this finding (Fig. 3E). Correlation analysis indicated that DNMT1 was closely associated with pyrimidine metabolism (Supplementary Figure S5A). Based on the results, DNMT1 was chosen as a representative m5C regulator. The relative expression levels of DNMT1 were examined in six cell lines, leading to the selection of the two with the highest DNMT1 expression, MHCC97H and Huh7 cell lines, for further experiments (Fig. 3F; Supplementary Figure S5B). To investigate potential mechanism of how DNMT1 regulates pyrimidine metabolism, DNMT1 was knocked down, with verification of the knockdown efficiency by RT-PCR and western blotting (Fig. 3G, H). Enzyme linked immunosorbent assay (ELISA) showed significant differences between the control and DNMT1-knockdown groups (Fig. 3I). It was thus concluded that DNMT1 plays a significant role in m5C modification. Furthermore, consistent with the previous hypothesis, knockdown of DNMT1 was found to reduce key enzymes expression in pyrimidine metabolism and the production of metabolites of pyrimidine metabolism (Fig. 3J–L; Supplementary Figure S5C, D). The cell densities of DNMT1-knockdown cells were markedly reduced compared with the controls (Supplementary Figure S5E, F). Furthermore, colony formation assay and transwell experiments showed that migration and invasion in both cell lines were significantly inhibited following DNMT1 knockdown (Fig. 3M, N; Supplementary Figure S5G). EdU experiment furthermore reinforced that knockdown of DNMT1 attenuates HCC proliferation (Supplementary Figure S5H). Together, these findings demonstrated that DNMT1 activated pyrimidine metabolism, thus promoting the migration and invasion of HCC cells.
Identification of five genes potentially associated with m5C modification by WGCNA analysisTo identify hub genes in the m5C clusters, Weighted Gene Co-expression Network Analysis (WGCNA) was performed (Fig. 4A–C; Supplementary Figure S6A–C, F). Hierarchical clustering of gene–gene modules (Fig. 4A) led to the identification of 19 distinct modules, shown by different colors in the figure (Fig. 4B, C). As shown in dendrogram and heatmap, the gray- and salmon-colored modules differed markedly from the others, and showed positive correlations with clusters cluster 2 and 3 (Fig. 4B, C), while the turquoise-colored module was primarily associated with cluster 1 (Fig. 4C). Fifty-three genes were identified in cluster 1 using a threshold of 0.8 for gene relativity. Furthermore, univariate Cox regression identified 50 genes that significantly affected survival. A protein–protein interaction (PPI) network of the m5C regulators and these 50 genes was then constructed using the STRING database (Fig. 4D). The PPI network showed that CDK1, RAD51, CCNB1, CENPA, and FOXM1 could potentially interact with DNMT1 (Fig. 4D). Analysis of differentially expressed genes, visualized by volcano and violin plots, showed that CDK1, RAD51, CCNB1, CENPA, and FOXM1 were upregulated in cluster 1, emphasizing the significance of these genes in cluster 1 (Supplementary Figure S6D, E). To visualize the significance of these 5 genes, scatter plots were used (Supplementary Figure S6F). Kaplan–Meier analysis of the effects of these genes on survival showed that higher expression was associated with poor prognosis (Fig. 4E). This finding might partially account for the poor survival outcomes observed in cluster 1. Given the previous demonstrations of the involvement of DNMT1 in both pyrimidine metabolism and m5C modification, its potential associations with key regulators of pyrimidine metabolism were investigated using Spearman correlations (Fig. 4F; Supplementary Figure S6G). The results indicated strong positive correlations between hub genes and pyrimidine metabolism. Together, these results indicate potential associations between DNMT1 and key genes involved in pyrimidine metabolism.
Fig. 4
Discovery of fundamental genes related to clusters with highest m5C levels. A Hierarchical clustering of genes, with different gene modules represented by different colors. B Clustering of different gene modules. C Heatmap showing correlations between gene modules and m5C clusters. The turquoise module is associated predominantly with cluster 1. D Protein–protein interaction network illustrating the hub genes associated with m5C regulators and five genes strongly linked to cluster 1 (unrelated interactions are omitted). E Kaplan–Meier curves comparing the high- and low-expression groups, shown in red and blue, respectively. F Spearman correlation analysis indicating significant associations between the expression of the five genes and DNMT1
DNMT1 modulates CDK1 via m5C modificationNext, the downstream targets of DNMT1 were explored, using m5C-RNA-immunoprecipitation sequencing (RIP-seq) to identify the characteristics of m5C modifications of DNMT1. As shown in the pie plot, the m5C peaks were decreased in the CDS, intronic, and intergenic regions following DNMT1 knockdown (Fig. 5A). Motif enrichment showed that DNMT1 knockdown altered the m5C sequence motif (Fig. 5B). These results indicated that DNMT1 played a significant role in gene modulation. To identify the downstream effector or effectors of DNMT1, RNA-seq was performed on DNMT1-knockdown cells, and differential analysis was conducted, leading to the identification of 405 downregulated genes compared with controls. The m5C-RIP-seq results also revealed 503 genes with m5C enrichment peaks. Venn diagrams were used to visualize genes located in the intersection of the m5C m5C-RIP-seq, RNA-seq, and WGCNA results, resulting in the identification of CDK1, KIF4A, and TPX2 (Fig. 5C). However, only the levels of CDK1 were observed to decrease significantly after DNMT1 knockdown, as seen in both the mRNA and protein levels (Fig. 5D–G). To further determine whether DNMT1 modulates CDK1 expression via m5C modification, the m5C peaks of CDK1 were plotted (Fig. 5H), showing a marked reduction in the signal intensities following DNMT1 knockdown (Fig. 5H, I). Moreover, CDK1 showed higher expression in HCC tissues and was associated with poor prognosis (Fig. 4E; Fig. 5J, K). Moreover, the proportion of CDK1 remaining after DNMT1 knockdown was reduced after Actinomycin D treatment (Fig. 5L, M). As further confirmation of the results, Spearman correlation analysis was conducted in two GEO datasets, GSE76427 and GSE54236, resulting in significant positive correlations (Fig. 5N, O). Therefore, the results demonstrated that DNMT1 modified CDK1 expression via m5C modifications, promoting the expression of CDK1.
Fig. 5
DNMT1 upregulates CDK1 via m5C modifications. A Comparison of the distribution of m5C modification sites in HCC cells between the control and DNMT1-knockdown groups. B m5C motifs on CDK1. C Venn diagram showing genes common to the m5C MeRIP-seq, RNA-seq, and WGCNA results. D, E Relative expression of CDK1, KIF4A, and TPX2 mRNA (D) and protein (E) levels after DNMT1 knockdown in two cell lines. F, G Western blot results (F) and RT-PCR measurements of the relative protein and mRNA expression, respectively, of CDK1 (G) showing that CDK1 levels were markedly reduced after DNMT1 knockdown. H m5C-IP results showing the m5C peaks in CDK1 mRNA. I Relative CDK1 fold enrichment after DNMT1 knockdown. J, K Comparison of CDK1 expression between tumor and normal tissues in the TCGA-LIHC cohort (J) and between normal and HCC tissues (K). L, M Analysis of CDK1 mRNA stability after DNMT1 knockdown and actinomycin D treatment. N, O Spearman correlations between DNMT1 and CDK1 expression in two GEO datasets GSE76427 (N) and GSE54236 (O). (*P < 0.05, ** P < 0.01, ***P < 0.001)
DNMT1 activates pyrimidine metabolism and promotes HCC progression by upregulating CDK1 in vitroTo investigate the role of CDK1 in HCC, the CDK1 gene was knocked down, resulting in significantly reduced colony formation on both the Huh7 and MHCC97H cell lines (Fig. 6A–C). In addition, migration and invasion of HCC cells were inhibited following CDK1 knockdown (Fig. 6D, E). Furthermore, EdU assays verified that CDK1 promoted HCC cell proliferation (Supplementary Figure S6A). To explore the association between CDK1 expression and survival outcomes, survival data from two GEO datasets, GSE76427 and GSE54236, were analyzed (Supplementary Figure S6B, C). Elevated CDK1 expression was linked to poorer clinical outcomes (Supplementary Figure S6B, C). The tumor doubling time was also significantly shorter in the high-CDK1 group compared with the low-CDK1 group (Supplementary Figure S6D). The result suggested that CDK1 may play a crucial role in tumor growth. Collectively, these findings suggest that CDK1 enhances HCC migration and invasion.
Fig. 6
DNMT1 indirectly promotes pyrimidine metabolism via upregulation of CDK1 expression, promoting HCC progression. A mRNA expression of CDK1 following CDK1 knockdown in two HCC cell lines, shown by RT-PCR. B Protein levels of CDK1 following CDK1 knockdown, shown by Western blotting. C Colony formation in the CDK1-knockdown groups vs. the control group in two HCC cell lines. D, E Transwell assay results showing the migration and invasion abilities of HCC cell lines. F, G RT-PCR results showing reduced expression of pyrimidine metabolism-related genes after knockdown of CDK1. H Rescue experiment showing increased CDK1 expression levels following DNMT1 overexpression. I Western blotting showing protein expression of CDK1 under different treatments. J Colony formation in DNMT1-overexpressing cells. K Transwell assay results showing migration and invasion abilities of DNMT1-overexpressing cells. (*P < 0.05, ** P < 0.01, ***P < 0.001)
Given that pyrimidine biosynthesis is fundamental to DNA synthesis which was closely associated with cell cycle (Fig. 3A), it was speculated that CDK1 was associated with pyrimidine metabolism. To explore whether CDK1 enhanced HCC malignancy via upregulation of pyrimidine metabolism in HCC, correlations between the expression of CDK1 and key enzymes in pyrimidine metabolism, as well as the GSVA scores of pyrimidine metabolism, were investigated (Supplementary Figure S7E, F). A significant correlation indicated that CDK1 potentially contributed to the promotion of pyrimidine metabolism. The relative expression of key enzymes involved in pyrimidine metabolism following the knockdown of CDK1 was substantially decreased (Fig. 6F, G). These results suggested that CDK1 regulates pyrimidine metabolism, promoting the migration and invasion of HCC cells, as well as being associated with poorer clinical prognosis. As it was previously found that DNMT1 induces dysregulated pyrimidine metabolism, it was hypothesized that DNMT1 upregulates CDK1 leading to the potential promotion of pyrimidine metabolism by the latter. To examine this hypothesis, knockdown and rescue experiments were performed (Fig. 6H, I). The inhibition of CDK1 was observed to significantly suppress colony formation, while overexpression of DNMT1 reversed the inhibitory effects of CDK1 knockdown (Fig. 6J). Furthermore, DNMT1 overexpression reversed the inhibitory effect of CDK1 knockdown on the malignant phenotype, promoting the migration and invasion of HCC cells (Fig. 6K). Together, these results demonstrated that DNMT1 modified CDK1 via m5C modification to upregulate CDK1, while CDK1 contributed to the promotion of pyrimidine metabolism to stimulate HCC progression.
Inhibition of the DNMT1/CDK1/pyrimidine metabolism axis suppresses HCC progression in vivoTo evaluate the potential of targeting the DNMT1/CDK1/pyrimidine metabolism axis in alleviating HCC progression, GSK3685032, a specific DNMT1 inhibitor, was used to treat HCC cells at doses of 0.1 and 1 μM. It was found that GSK3685032 treatment led to reduced cell proliferation and colony formation (Fig. 7A–C; Supplementary Figure S7A). The inhibitory effect was dose-dependent, with greater inhibition seen at higher doses (Fig. 7A–C; Supplementary Figure S8A). Moreover, the tumorigenic behavior of HCC cells was suppressed by the inhibitor in a dose-dependent manner (Fig. 7D, E). The specific CDK1 inhibitor Ro-3306 was also used to treat HCC cells, after which the relative expression levels of key enzymes involved in pyrimidine metabolism were measured. This indicated significantly reduced expression of these enzymes in the presence of both GSK36 and Ro-3306 (Fig. 7F, G). Thus, treatment with GSK36 and Ro-3306 could downregulate pyrimidine metabolism in HCC cells. BAY2402234 is an inhibitor of pyrimidine metabolism. In vivo experiments in mice showed that the GSK36, Ro-3306, and BAY-treated groups showed significantly lower luminescence in tumors compared with the control group (Fig. 7H). Following the use of specific inhibitor of DNMT1, m5C levels were detected, and it was found that m5C reduced significantly after GSK36 treatment. Additionally, H&E and Ki-67 staining indicated that the GSK36, Ro-3306, and BAY24 treatment groups displayed reduced tumor cell densities relative to the control group (Fig. 7I). Together, these results demonstrated that targeting the DNMT1/CDK1/pyrimidine metabolism axis in HCC suppressed HCC progression.
Fig. 7
Inhibition of DNMT1, CDK1, and pyrimidine metabolism reduces malignant progression of HCC. A, B Effects of the DNMT1 inhibitor (GSK3685032) on cell growth. Higher concentrations had a greater inhibitory impact on cell proliferation. C In vitro cell culture showing reduced colony formation after incubation with varying concentrations of the DNMT1 inhibitor in two cell lines. D, E Transwell assay results showing reduced migration and invasion after inhibition of DNMT1. F, G RT-PCR measurements of the relative expression of pyrimidine metabolism-related genes after inhibition of DNMT1 in two cell lines. H Treatment of MHCC97H mouse models with the DNMT1 inhibitor (GSK3685032), CDK1 inhibitor (Ro-3306), and pyrimidine metabolism inhibitor (BAY2402234). Luminescence intensities of tumors on days 7 and 35, indicating inhibitory effects on HCC. I H&E and Ki-67 staining of tumor tissues. Scale bar, 25 μm. (*P < 0.05, **P < 0.01, ***P < 0.001)
Comments (0)