Patient samples and clinical data collection
Eleven HCC tissues and ANTs were collected in the Shanghai Eastern Hepatobiliary Surgery Hospital. The inclusion criteria were patients who were diagnosed with HCC and received curative surgery. All samples underwent strict histopathological evaluation to ensure diagnostic accuracy and ensured the HCC tissues contained more than 80% tumor cells. The exclusion criteria were patients who received preoperative treatment for HCC or had a history of other cancers. All tissues were collected and immediately frozen in liquid nitrogen and stored at − 80 °C until DNA was extracted. The samples were then used for Illumina HM450K methylation chip microarray analysis. Our study was conducted in strict compliance with the Declaration of Helsinki (as revised in 2013) and was approved by the Ethics Committees of the hospitals. Informed consent was obtained from all included patients.
DNA extraction and bisulfite conversion
A QIAamp DNAMini Kit (QIAGEN, Hilden, Germany) was used to extract genomic DNA from ≥ 25 mg primary cancerous tissues and ANTs. The concentrations and quality of all DNA samples were detected using a Thermo NanoDrop 2000 spectrophotometer. Then, 400–500 ng of DNA was taken for bisulfite treatment using an EpiTect Fast DNA Bisulfite Kit (QIAGEN, Hilden, Germany).
Methylation microarray analysis
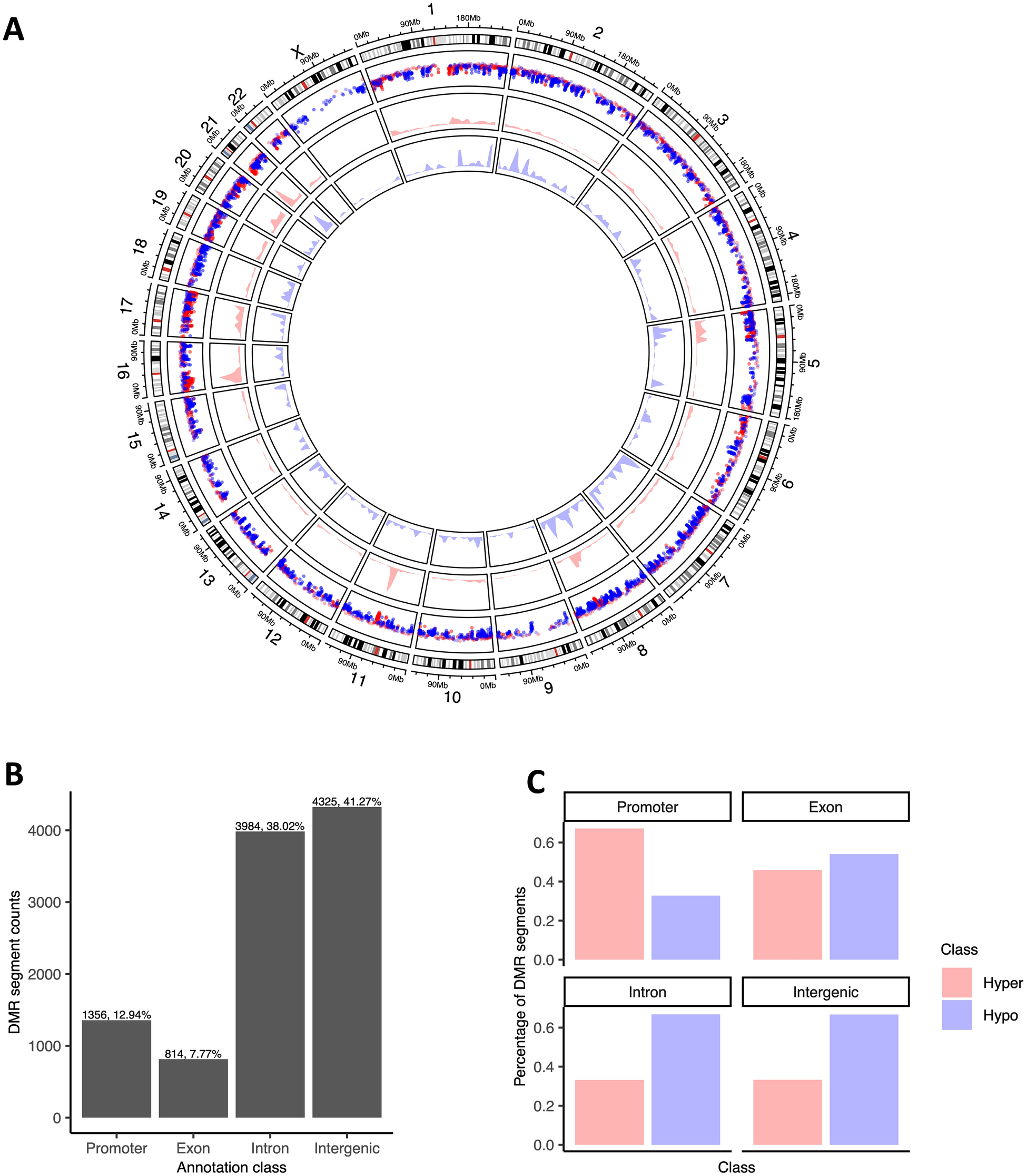
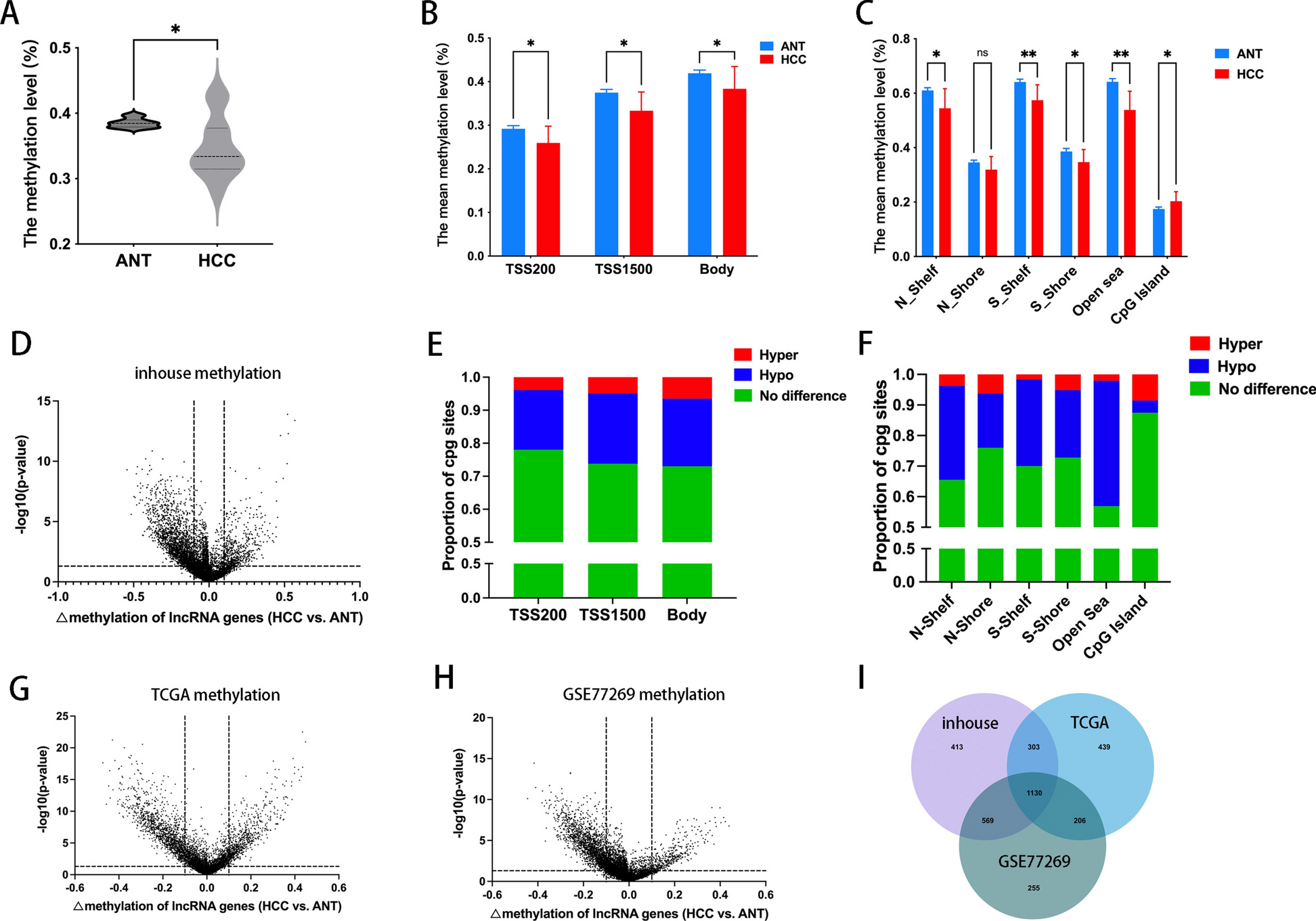
500 ng bisulfite-treated DNA was employed for genome-wide methylation analysis via the Infinium HM450 BeadChip (Illumina, San Diego, CA, USA). Subsequent normalization and filtration of the raw methylation matrices were executed in R (v3.4.3; https://www.r-project.org/) utilizing the Chip Analysis Methylation Pipeline (ChAMP) package [10]. The microarray results were expressed as the percentage of methylation signal intensity at different sites by β value. β = M/(M + U + O), where U represents the unmethylated signal intensity, M represents the methylated signal intensity, and O is the offset to prevent the occurrence of zero as a denominator. The Infinium HM450 BeadChip contained 485577 CpG sites. After annotation of the BeadChip according to the ENCODE database (human genome GRCh38, version 42), a total of 11928 CpG sites were corresponded to lncRNA genes and selected for further analysis. Differentially methylated CpG sites that corresponded to lncRNA genes were further screened from these 11928 CpG sites by calculating the difference in methylation level of each CpG site between HCC tissues and ANTs. The criteria for the difference were set as: |delta methylation difference|> 10% and P < 0.05. The volcano plot was drawn using GraphPad Prism 9. The heat map was drawn using the R package “heatmap.”
Online datasets and samples
Online HCC methylation datasets from the Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) database (GSE77269) were used for conjoint analysis, along with our in-house methylation microarray data. A total of 20 HCC tissues and ANTs were included in the GSE77269 database. In total, 369 HCC tissues and 49 ANTS were included in the TCGA database. These two datasets used Illumina HM450 BeadChip, and methylation differences were analyzed in the same way as our in-house database. For different lncRNA gene expressions in the TCGA database, the limma package in the R software was utilized at the cutoff criteria of P < 0.05 and fold change (FC) > 2.0. The heat map of differentially expressed lncRNA genes was drawn using the R package “heatmap.”
Construction of the prognostic model
LncRNA genes with both methylation and expression differences were screened for prognostic model construction. Since one lncRNA gene often corresponded to multiple CpG sites, we calculated the mean methylation value of all CpG sites that correspond to a single lncRNA gene as the methylation level of that lncRNA gene. The prognostic model was constructed based on the methylation levels of these lncRNA genes and prognostic information of HCC patients in the TCGA database. Patients without lncRNA methylation data or prognostic information were excluded. Patients were allocated to the training and validation groups in a 7:3 ratio. LASSO regression analysis was applied to the training group to reduce the likelihood of overfitting and to narrow down the lncRNA candidates [11]. Next, a multivariate Cox regression model was used to identify lncRNA genes most related to survival, from which the risk models were constructed. The risk score for each HCC patient was calculated using the formula below: Risk score = Coef lncRNA1 × lncRNA1 methylation + Coef lncRNA2 × lncRNA2 methylation + ··· + Coef lncRNAn × lncRNAn methylation. Coef represented the coefficient value of the corresponding lncRNA.
Evaluation of the prognostic model
The prognostic model was evaluated in both the training and validation groups. Patients were categorized into low-risk and high-risk groups based on the median risk score. Then, principal component analysis (PCA) was used to explore potential differences between high-risk and low-risk groups [12]. The model's performance and prognostic ability were evaluated using time-dependent ROC analyses and Kaplan–Meier (K–M) log-rank tests, via the R packages “timeROC” and “survival” [13, 14]. An area under the curve (AUC) of greater than 0.7 was considered predictive.
Cell lines and reagents
The human hepatocellular carcinoma cell lines HepG2 and Huh7 and tool cell HEK293T were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in DMEM (Gibco, USA) containing 10% fetal bovine serum (FBS, Gibco) at 37 °C in humidified air with 5% CO2. Decitabine (NSC-127716) was purchased from MCE.
Decitabine treatment
HCC cells were pre-cultured to 10–25% confluence and then cultured with decitabine (DAC, Targetmol)-containing medium at different concentrations (0 μM, 5 μM, and 10 μM) for three days. DAC was diluted in dimethylsulfoxide (DMSO, Sigma). The medium was refreshed every three days.
Lentiviral vector construction and transfection
Incorporation of sh-LINC00942-1, sh-LINC00942-2, and scrambled shRNA into the pLKO.1 plasmid was conducted, followed by transient transfection of HEK293T cells with lentiviral vectors, packaging, and envelope plasmids MD2G and PAX2 using Lipofectamine™ 3000 transfection reagent (L3000015; Thermo Fisher, Waltham, MA, USA) for lentivirus supernatant production. Viral supernatants were subsequently harvested, filtered through a 0.22 μM filter (Millipore, Burlington, MA, USA), and ultracentrifuged. After lentivirus transfection for 48 h, HCC cells were subjected to puromycin selection at 1 μg/mL. The short hairpin RNA sequences for LINC00942 knockdown were as follows:
sh-LINC00942-1: GCTGGCTCCTGCTTGGAAACT;
sh-LINC00942-2: GCTTGGAATTGATGACGTTTC.
RNA extraction and quantitative real-time PCR
RNA extraction was performed using the RNA-Quick Purification Kit (AG21023, Accurate Biology). Reverse transcription was executed following the protocol of the Eco M-MLV RT Premix kit (AG11706, Accurate Biology). The target gene expression was normalized to the endogenous control gene GAPDH. RT-qPCR was performed using the SYBR Green Premix Pro Tag HS qPCR kit (AG11701, Accurate Biology) on the QuantStudio 1 machine (Applied Biosystems, Thermo Fisher Scientific, USA). The RT-qPCR primer sequences were as follows:
LINC00942-F: GGTGTCTGCGGGAAACAGTAC,
LINC00942-R: GAACAAAGAGTCAGGTTGTGTGG;
GAPDH-F: CTCTGCTCCTCCTGTTCGAC,
GAPDH-R: ACCAAATCCGTTGACTCCGA.
Methylation-specific PCR (MSP)
Firstly, cellular genomic DNA was collected using the Universal Genomic DNA Purification Mini Spin Kit (D0063, Beyotime, Shanghai, China) following the provided instructions. Subsequently, the EpiArt® DNA Methylation Bisulfite Kit (EM101, Vazyme, Nanjing, China) was employed to perform the sulfite conversion of DNA and to purify it. To perform the sulfite conversion of DNA, 20 μl of DNA (approximately 100 pg–2 μg) was mixed with 130 μl of CT Conversion Mix. The mixture was then placed in a PCR apparatus and processed according to the program settings. Following this, the product was purified. To collect the purified DNA filtrate, 10–20 μl of E-Elution Buffer was added to the center of the adsorption column membrane. The cells were allowed to stand for 1–2 min at room temperature and centrifuged at 12,000 rpm for 2 min, and the purified DNA filtrate was collected. Subsequently, primers targeting methylated and unmethylated sequences were designed using MethPrimer (http://www.urogene.org/methprimer/) and amplified through PCR. The methylation status of DNA sequences complementary to the primers was determined by agarose gel electrophoresis. The specific primers targeted methylated and unmethylated sequences were as follows:
LINC00942-M-F: TAATTTTTTTTGTTGTTTTTAGCGT,
LINC00942-M-R: TATAAAAATTTCTCAAACATTCGAT;
LINC00942-U-F: TTTTAATTTTTTTTGTTGTTTTTAGTGT,
LINC00942-U-R: TATAAAAATTTCTCAAACATTCAAT.
Cell viability and proliferation assay
A total of 1000 HCC cells per well were planted into a 96-well plate and allowed to attach for 24 h. Then, 100 mL of fresh medium containing 10% CCK8 solution (MA0218, Meilunbio, China) was added at 1, 2, 3, 4, and 5 days after planking, and the 450-nm absorbance was detected following incubation for 2 h at 37 °C using a spectrophotometer (Multiskan Spectrum 1500, Thermo, USA).
Transwell cell migration and invasion assay
HepG2 and Huh7 cells were seeded into the upper compartments of 24-well plates at a density of 30,000 cells per well, using 100 μl of serum-free DMEM. In the lower compartment of the plate, 0.6 ml of DMEM supplemented with 10% FBS was added as a chemoattractant. The plates were then incubated at 37 °C for 24 h. Following the incubation period, the cells on the lower side of the insert membrane were fixed with 4% paraformaldehyde for 30 min and then stained with 1% crystal violet for an additional 20 min. The inserts were rinsed with PBS for several seconds to remove excess dye and then allowed to air dry completely. Pictures of the lower side of the filter were captured under a microscope. For the transwell invasion assay, 50 μl of 10% ECM gel (40183ES08, Yeason, China) was added to the upper compartment of the insert, and the plates were incubated at 37 °C for 2 h to allow the liquid ECM gel to solidify. The remaining steps followed the protocol described above.
Cell cycle flow cytometry
A total of 5 × 105 cells were harvested and washed twice with PBS. Subsequently, each group was treated with 1 mL of DNA staining solution containing 10 μL of permeabilization solution (Cell Cycle Staining Kit, CCS012, MultiSciences, Hangzhou, China) and incubated for 30 min in the dark at room temperature. The samples were then subjected to flow cytometry analysis using a BD LSRFortessa instrument (New Jersey, NJ, USA). The results were analyzed using the “FlowJo” software.
Cell apoptosis flow cytometry
Cells inoculated in 6-well plates were collected (about 1–10 × 105 cells, including those in the culture supernatant) and washed twice with PBS. Cells were resuspended in 500 μl of 1 × Binding Buffer. 5 μl Annexin V-APC and 10 μl PI were added to each tube (Annexin V-APC/PI Apoptosis Kit, AT107, MultiSciences, Hangzhou, China). After gently vortexing and thorough mixing, the mixture was incubated at room temperature in the dark for 5–10 min. Following this incubation period, flow cytometry analysis was carried out, and the results were analyzed using the “FlowJo” software.
Western blotting and antibodies
Cell protein extraction was performed using RIPA buffer (Fude Biotech, China) with protease inhibitors added. Protein concentrations were measured with a BCA Protein Assay Kit (Meilunbio, China). Then, 10–20 μg of protein was loaded onto a sodium dodecyl sulfate–polyacrylamide gel for electrophoresis and transferred to a 0.22- or 0.45-µm-pore-sized PVDF membrane. The PVDF membranes were blocked with 5% skim milk for 2 h and then incubated with specific primary antibodies overnight at 4 °C. The primary antibodies used were as follows: GAPDH (AC002, Abclonal, Wuhan, China), α-tubulin (ab7291, Abcam, Cambridge, UK), Cell Cycle (CDK2, CDK6, Cyclin B1, Cyclin D1) Antibody Sampler Panel (ab228528, Abcam, Cambridge, UK), Pro-Apoptosis BCL-Family (Bad, Bax, Bcl-2), Antibody Sampler Panel (ab228527, Abcam, Cambridge, UK), and Active Caspase-3 Antibody (SR01-02, Thermo Fisher, MA, USA). Afterward, the membranes were incubated with the appropriate secondary antibodies for 2 h at room temperature. Finally, the protein bands were visualized using an enhanced chemiluminescence (ECL) western blotting substrate (Fude Biotech, China).
Establishment of hepatoma xenograft model
The male BALB/c nude mice (n = 5, 4 weeks old, weighing ~ 20 g) were purchased from the Shanghai Laboratory Animal Center and fed in a pathogen-free environment at the Laboratory Animal Research Center of SRRSH. All animal experiments were strictly designed and executed according to the guide approved by the Committee on the Use of Live Animals in Teaching and Research at SRRSH. We injected 1 × 107 control or sh-LINC00942-treated HepG2 cells in 200 μL PBS into the groin of nude mice to form murine subcutaneous tumors. The tumor volumes were measured every two days, which were calculated as follows: V tumor = L × W2/2, where L and W were, respectively, the length and width of the tumor. The mice were killed 15 days after injection, and the tumors were isolated, weighed, and photographed.
Hematoxylin–Eosin (H&E) staining
H&E staining was used to observe the histological structure of subcutaneous tumors in mice. Tumors were excised and fixed overnight in 4% paraformaldehyde. Following fixation, the tissues were dehydrated, embedded in paraffin, and cut into 5-µm-thick sections for preservation. After deparaffinization and gradient hydration, the paraffin sections were stained with hematoxylin for 2 min, washed with PBS, and then stained with eosin for 1 min. The stained sections were dehydrated using a gradient of alcohol and preserved with neutral gum sealing. Images were captured under a light microscope for observation.
Immunohistochemistry (IHC)
IHC was used to evaluate Ki-67 expression in tumoral hepatic tissues with and without LINC00942 knockdown. Following paraffin embedding, 5-mm sections of tissue samples were prepared. The sections were first deparaffinized and rehydrated. For antigen retrieval, they were immersed in 10 mM citrate buffer (pH 6.0) and heated in a microwave oven for 10 min. Endogenous peroxidase activity was then inhibited using 3% hydrogen peroxide for 10 min. To block non-specific binding, the sections were treated with 10% normal goat serum for 30 min. Subsequently, the sections were incubated overnight at 4 °C with an anti-Ki-67 antibody (1:500, A20018, Abclonal, Wuhan, China). Following this, they were incubated with a secondary antibody, and Ki-67 expression in the tissues was evaluated via microscopy after diaminobenzidine and hematoxylin staining.
Statistical analysis
SPSS version 21.0, GraphPad Prism 9, and RStudio were used to conduct all statistical analyses. Statistical significance between the two groups was calculated with a two-sided Student’s t-test. Variances among multiple groups were analyzed by one-way ANOVA. A difference in methylation level was defined as the absolute value of the difference in methylation level between HCC tissues and ANTs greater than 10% and a P value less than 0.05. The correlations between methylation levels and methylation level of lncRNA genes were assessed by Pearson correlation analysis. Statistical significance was defined as a p value < 0.05.



Comments (0)