Cell lines, antibodies, and chemicals
All cell lines were obtained from Cell Bank of Shanghai, Institutes for Biological Sciences, China and negative for mycoplasma infection. Cells were cultured in DMEM medium or RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in a humidified atmosphere with 5% CO2. Antibodies against ARID1A, HP-1a, STAT1, H3, acetyl-H3K9 and acetyl-H3K27 were purchased from Cell Signaling Technology (Danvers, MA, USA), when used for western blotting, immunofluorescence or immunohistochemistry antibodies were diluted according to manufacturer’s instructions. The dilution rate for each antibody was given in Table 1. For staining immune cells in tumor tissues in flow cytometry analysis, PE-anti-CD45, APC Cy7-anti-CD3, FITC-anti-CD4, PerCP Cy5.5-anti-IFNg and PE Cy7-anti-TNFa antibodies were purchased from BioLegend (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA). HDAC inhibitors (HDACi: LBH589 and TSA) were purchased from Selleck chem (Selleck Chem, Houston, TX, United States). PD-L1 mAb was purchased from BioXcell (Lebanon, NH, United States). Concentration of LBH589 used in vitro was between 10 and 200 nmol/L, when used in vivo the concentration was 10 mg/kg.
Table 1 Primary antibodies dilution rateGeneration of stable cells by lentiviral infection
ARID1A knockdown lentivirus was purchased from Genechem (Genechem, Shanghai, PR China). To select cells that ARID1A were stably knockdown, cells were plated at subconfluent densities and infected with a cocktail of 1 ml of virus-containing medium, 1 ml of regular medium and 8 μg/ml polybrene, and then selected in 1 μg/ml of puromycin (Sigma-Aldrich, Louis, MO, USA) 48 h after lentivirus infection.
Small interfering RNA (siRNA)
siARID1A was purchased from GenePharma siRNA. Transfection was performed at a concentration of 20 nM siRNA. GenePharma siRNA sequence was in Table 2.
Table 2 PCR, ChIP-PCR primer sequences, and siRNA sequences used in this studyBioinformatics analysis
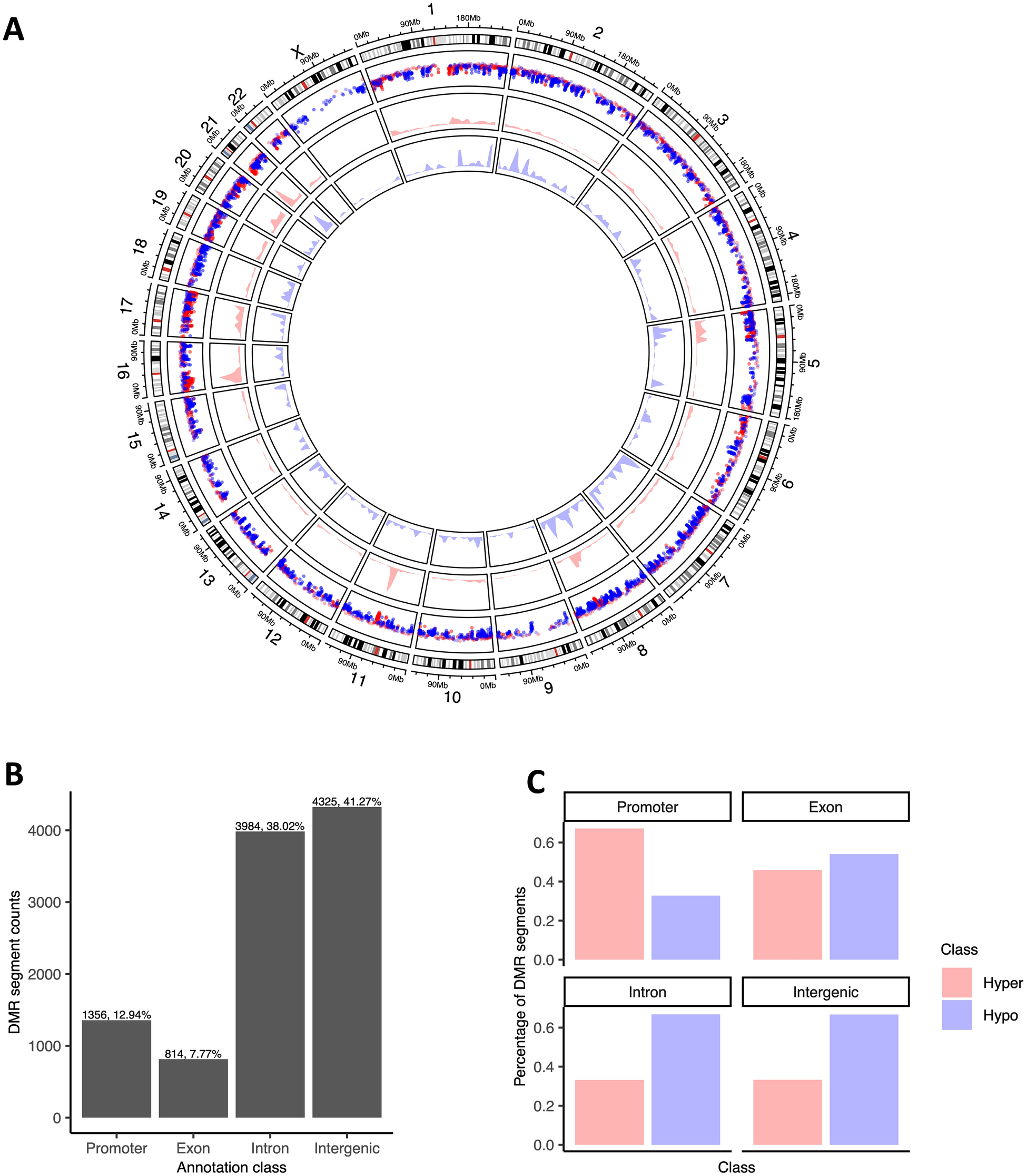
The GEO: GSE15460 dataset was downloaded from the Gene Expression Omnibus (GEO). Spearman correlation analysis between the expression of CXCL9, CXCL10 and ARID1A in human gastric cancer tissues was conducted using RNA-seq data from GSE15460 dataset (n = 329).
Total RNA extraction and cDNA synthesis
Total RNA was extracted from cells using TRIzol Reagent (Takara, Kusatsu, Japan) following the manufacturer’s instructions. After assessing quality and quantity, samples were then stored at − 80 °C. The extracted RNA was reverse-transcribed into cDNA using PrimeScript™ RT Master Mix (Takara, Kusatsu, Japan). The resulting cDNA was stored at − 20 °C for further analysis.
Quantitative real-time PCR (qRT-PCR)
Gene expression levels were quantified using qRT-PCR with gene-specific primers and One Step SYBR PrimeScript RT-PCR Kit II (Takara, Kusatsu, Japan). The qRT-PCR reaction was following the manufacturer’s instructions. Expression levels were normalized to β-actin, and relative quantification was performed using the 2^-ΔΔCt method. Primers used was in Table 2.
Protein extraction
The cells were suspended in RIPA buffer (Sigma-Aldrich, Louis, MO, USA). Protease inhibitor cocktail (1%, Thermo Fisher Scientific, Waltham, MA, USA) was also added to the mixture. The lysate was collected by centrifugation at 12,000 rpm at 4 °C for 15 min. The supernatant was transferred to a new tube, after using BCA protein quantification assay to determine concentration, supernatant was then mixed with loading buffer (Sigma-Aldrich, Louis, MO, USA) and boiled at 95 °C for 10 min.
Histone extraction was performed differently, cells were resuspended in hypotonic lysis buffer with protease inhibitor cocktail (1%). Nuclei were then isolated by centrifugation, resuspended in strong lysis buffer to extract histone, and histones were collected by centrifugation. After histone extraction, quantification was performed by same way using BCA protein quantification assay.
Western blotting analysis
Protein samples (0.5–20 μg) were subjected to SDS-PAGE gel electrophoresis, and transferred to PVDF membranes. After blocking with 5% non-fat milk in Tris-buffered saline with Tween (TBST) the membranes were incubated with specific antibodies overnight at 4 °C with gentle agitation. After washing, membranes were incubated with HRP-conjugated secondary antibodies. Protein bands were visualized using chemiluminescent substrates.
Immunohistochemistry (IHC)
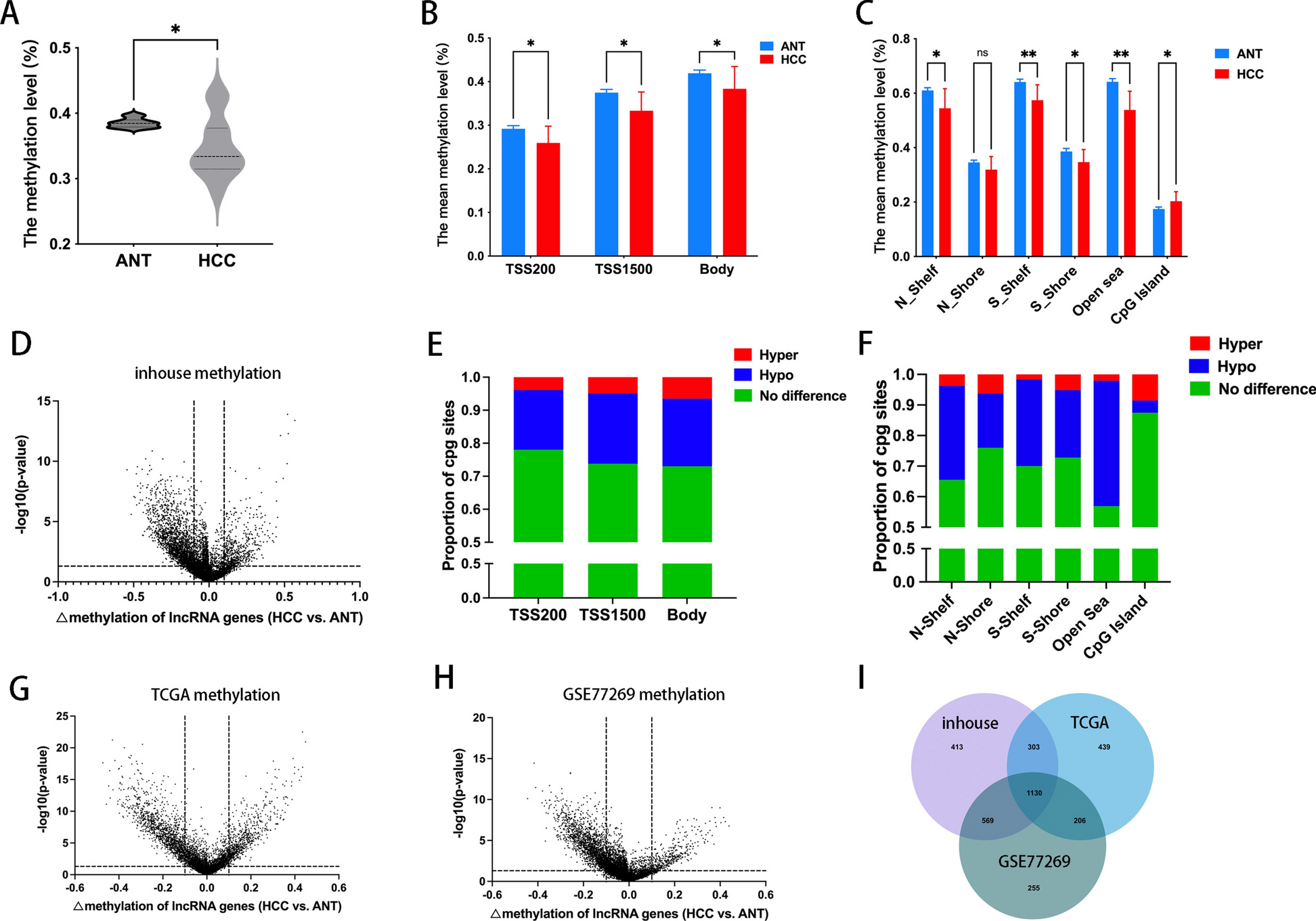
The sections of human gastric cancer tissues (n = 90) were dewaxed and rehydrated. Antigen retrieval was performed using citrate buffer, and endogenous peroxidase activity was blocked with 3% H2O2. The sections were incubated with blocking buffer (5% BSA) for 1 h at room temperature. Primary antibodies were incubated with the sections overnight at 4 °C. After washing three times with PBS for 5 min, the sections were incubated with secondary antibodies for 1 h. Hematoxylin was used to counterstain the tissue for visualization of the architecture, and the sections were mounted for microscopic examination. Spearman correlation between CD8-positive cells and ARID1A expression scores were analyzed using ImageJ (NIH, Bethesda, MD, USA).
Immunofluorescence
The sections were incubated with blocking buffer (5% BSA) for 1 h at room temperature, washed with blocking buffer. Diluted primary antibodies were incubated with the sections overnight at 4 °C. The primary antibodies were washed three times with PBS for 5 min, and the diluted secondary antibodies were dropped on the sections at room temperature for 2 h. The secondary antibodies were also washed three times with PBS for 5 min. After dropping the mounting medium with DAPI, the sections were covered with cover slides, and the edges were sealed. After observation under a fluorescence microscope, the sections were stored at 4 °C. The fluorescent intensity was analyzed using ImageJ (NIH, Bethesda, MD, USA).
Chromatin immunoprecipitation (ChIP)
Chromatin was cross-linked with 1% formaldehyde for 10 min and fragmented by sonication. Chromatin DNA was extracted using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, Danvers, MA, USA) following the protocol provided by the manufacturer. DNA was immunoprecipitated using specific antibodies. ChIP signals were quantified by qRT-PCR analysis with One Step SYBR PrimeScript RT-PCR Kit II (Takara, Kusatsu, Japan). Values obtained for immunoprecipitated samples (percent (%) input DNA) were normalized to values for respective normal IgG. The specific primer pairs for the CXCL9 promoter region and CXCL10 promoter region, respectively, are described in Table 1.
ChIP-sequencing was performed by BGI (BGI-Shenzhen, Shenzhen, PR China.). After DNA-end repair, 3′-dA overhang and ligation of methylated sequencing adaptor, samples were undergoing PCR amplification and size selection. Sequencing was performed after library qualification.
Chromatin accessibility analysis
Peaks were called using MACS2 and quantitated across samples using Seqmonk generating reads per kilobase per million mapped reads (RPKM). Peaks were annotated to genomic features and nearest promoter via the Homer annotate function. The reads from replicates for each peak were averaged and the peaks were ranked based on the difference between the average counts among conditions. The “pheatmap” R package was used to plot the top 1000 peaks heatmap.
DNA accessibility by MNase digestions
Chromatin DNA samples were prepared as method described above. Then samples were digested by MNase with different concentration. Transmission electron microscopy analysis was performed by Servicebio (Servicebio-Wuhan, Wuhan, PR China.). DNA accessibility was measured by agarose gel electrophoresis.
Transmission electron microscopy
Electron microscopy cells seeded at 1.105 per cm2 on the soft matrices were cultured for 24 h and fixed in 2% PFA-2% glutaraldehyde in 50 mM cacodylate buffer at pH 7.4 for 2 h and then fixed in 1% osmium tetroxide in 125 mM cacodylate for 30 min. After resin polymerization, resin blocks were cut to 60–80 nm thickness on the ultramicrotome, and the tissues were fished out onto the 150 meshes cuprum grids with formvar film. After 2% uranium acetate saturated alcohol solution stained for 8 min avoiding light, sections then rinsed in 70% ethanol and ultrapure water. Then sections were stained in 2.6% lead citrate solutions for 8 min avoiding CO2, then rinsed with ultrapure water. After dried by the filer paper, the cuprum grids were put into the grids board and dried overnight at room temperature. The cuprum grids are observed under HITACHI HT7800 (HITACHI, Tokyo, Japan) and take images.
Gastric cancer patient survival data analysis
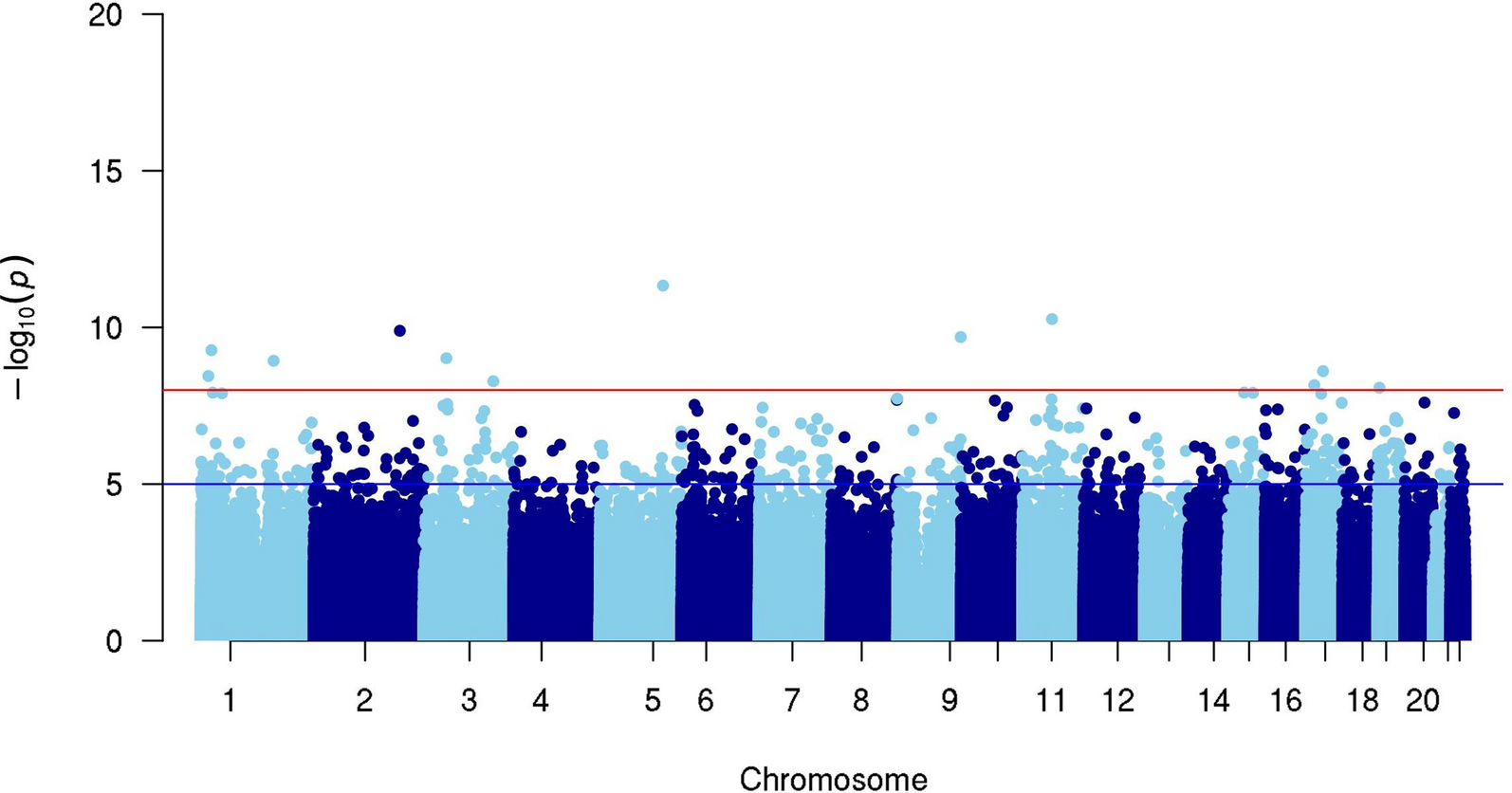
RNA-seq and patient survival data of gastric cancer patients in GEO dataset GSE62254 was downloaded from website. ARID1A low and ARID1A high group were divided based on expression level of ARID1A. The “survival” R package was applied to plot Kaplan–Meier curves and to compute the associated log-rank p-value.
Animal model
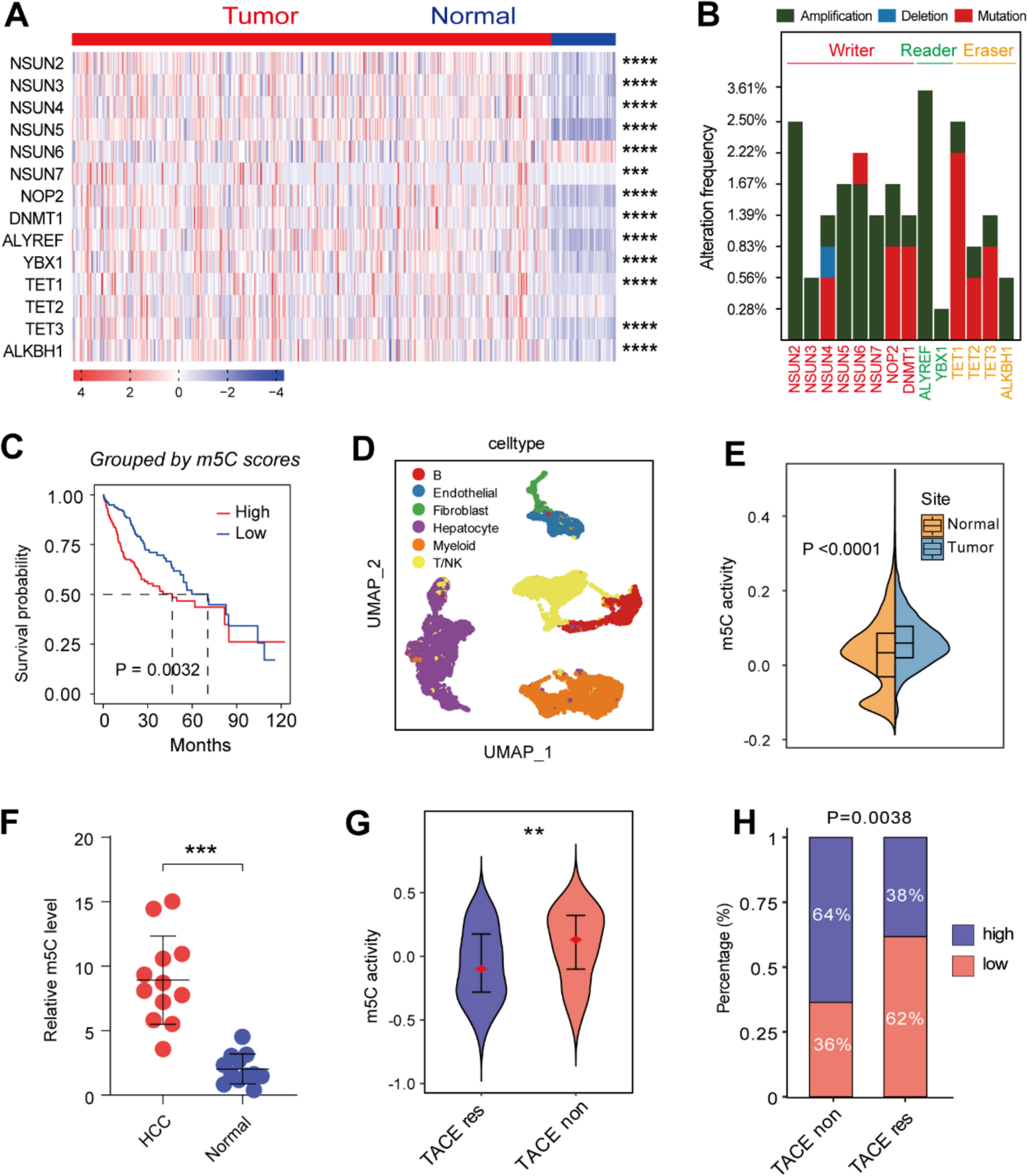
All experiments were approved and performed according to the guidelines of the Ethical Committee of Union Hospital, Huazhong University of Science and Technology. 615 mice (6 weeks old) were purchased from Shulaibao (Shulaibao, Wuhan, PR China). The MFC or MFC/ ARID1A knockdown cells were inoculated s.c. (5 × 105 cells/100 μL in PBS/mice) into the flanks of anesthetized mice. Mice in each group intra-peritoneally received LBH589 in 10% DMSO solution (10 mg/kg/day) or vehicle and anti-PD-L1 antibody (100 μg/day) (BioXcell, Lebanon, NH, United States) twice a week. The tumor volume was assessed every two days starting from day 3 after tumor inoculation.
Flow cytometry
Subcutaneous tumors were entirely separated from tumor-bearing mice, then were all pestled through 40 μm mesh. Tumor cells were removed by centrifuging using FACS buffer. Single cell suspensions of each sample were then used in following flow cytometry analysis after activation. Flow cytometry was performed using fluorescence-conjugated antibodies and their controls followed by species-specific conjugate using a FACS CantoII flow cytometer (BD Biosciences, San Jose, CA, USA). Positive cells in percentage (%) were calculated as follows: Surface expression in percent obtained with the specific antibody—surface expression in percent obtained with isotype control. T cells were preselected by CD45+ and CD3+. All flow cytometry data were analyzed using FlowJo software (Treestar Software, Ashland, OR, United States).
Clinical data collection
A total of 90 eligible patients were finally enrolled in this study. Study collected data from locally advanced gastric cancer patients who underwent surgery at the Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, between April 2019 and January 2020. We included the patients on the basis of the following inclusion criteria: (1) gastric cancer confirmed in histological biopsy; (2) clinical TNM stages II, III, and IVa; (3) radical gastrectomy combined with D2 lymph node dissection; and (4) complete clinicopathological data.
Statistical analysis
Significance was determined using GraphPad Prism software (GraphPad Software, San Diego, CA, United States). All statistical analyses were based on data obtained from a minimum of three independent experiments. For comparisons involving more than three means, we utilized one- or two-way ANOVA with Bonferroni correction. Two means were compared using the unpaired Student’s t-test. A significance level of p < 0.05 was considered statistically significant.



Comments (0)