The clinical manifestations evident in our cohort of eight children are like those described in the literature, with severe failure to thrive, cholestatic jaundice, and gross motor delay being the most frequent symptoms.
All patients in our cohort were homozygous for a severe MPV17 pathogenic variant, expected to result in no functional protein. It is therefore possible that cases with milder presentations caused by variants that only reduce or alter the MPV17 channel’s functionality, rather than result in absence of the channel, may present differently. This could explain the discrepancies in severity of the various clinical manifestations and the inconsistent imaging presentations documented in the existing literature [9]. No information was available on the pathogenic variant affecting the siblings with reticular abnormalities described previously [10].
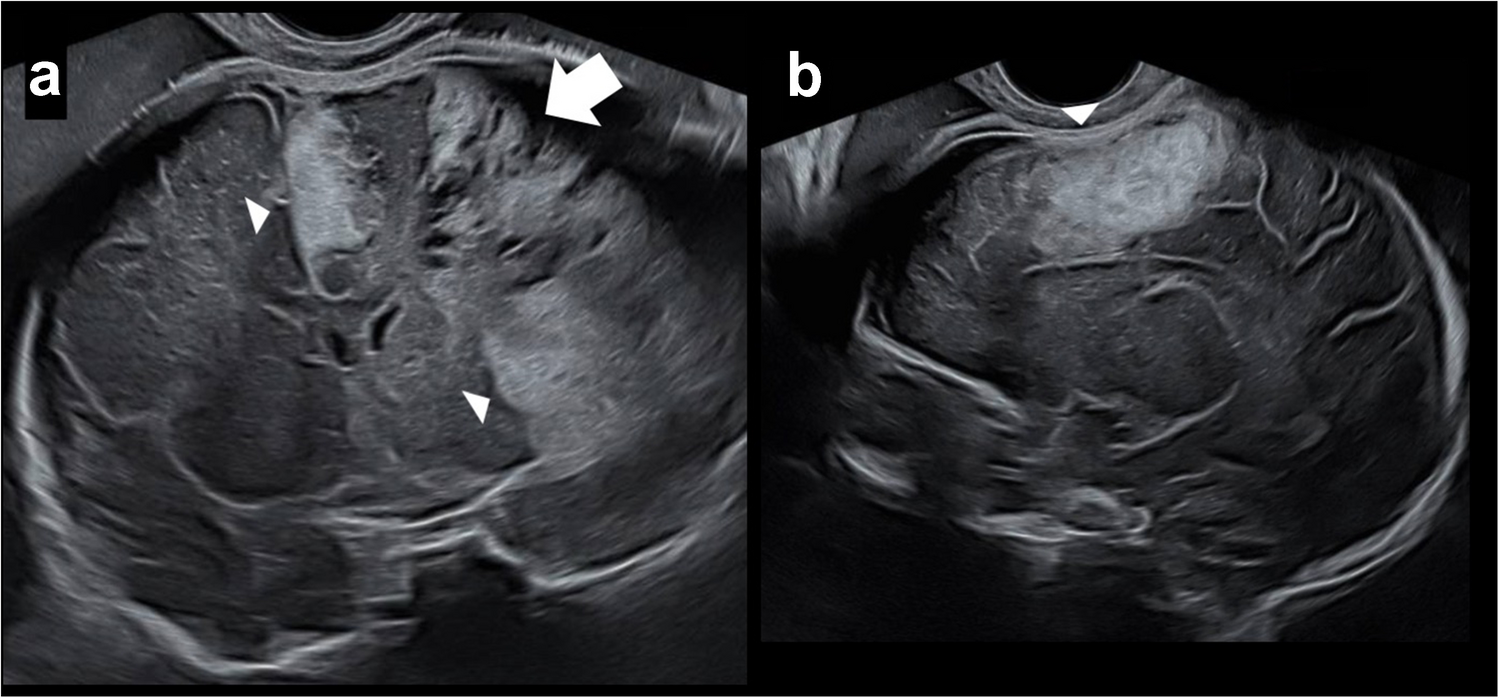
To our knowledge, the only cases published in the radiological literature are those of a sibling pair and three children from Saudi Arabia with variant-undisclosed MPV17 who showed identical abnormalities of the reticulospinal tracts and reticular formation on 1.5-T MRI [10]. Our findings strongly corroborated the hypothesis that this anatomical subsite is consistently affected in MPV17-related mitochondrial DNA depletion syndrome patients, with seven out of eight of the patients in our series (and all encephalopathic patients) showing the same MRI abnormality.
The reticular formation is made up of a net-like structure of various brainstem nuclei and neurons and covers an expansive portion of the brainstem, beginning in the mesencephalon, extending caudally through the medulla oblongata, and projecting into the superior cervical spinal cord segments. The reticular formation does not have any distinct cytoarchitectural boundaries and is dispersed throughout the brainstem as a network of interconnected neurons with many projections cranially to subcortical and cortical brain structures as well as caudally to the spinal cord [14]. The reticulospinal tract is a descending tract present in the white matter of the spinal cord, originating in the reticular formation. It consists of bundles of axons that carry information from the reticular formation in the brainstem to the peripheral body parts, being primarily responsible for locomotion and postural control [15]. Since these structures reside at the craniocervical junction, which is located at the inferior edge of the field of view on standard brain MRIs, they may potentially be overlooked. For this reason, knowledge of this anatomical area and recognition of its relevance in MPV17-related mitochondrial DNA depletion syndrome are essential in directing the imaging search. With higher magnetic field imaging at 3-T becoming increasingly available, it is expected that signal changes to the reticular formation should be more readily observed.
In addition to our study reinforcing the significance of the reticular formation and reticulospinal tracts in MPV17-related mitochondrial DNA depletion syndrome, it further characterises the topography, frequency, and extent of extra-reticular findings, including a predilection for the basal ganglia, a tendency to produce severe leukoencephalopathy, and frequent involvement of the corticospinal tracts. We establish that restricted diffusion occurs almost always at extra-reticular sites of involvement but infrequently in the reticular formation. Novel areas of interest, not previously described in the literature, are the perirolandic cortices and hippocampal formations, both regions of high mitochondrial activity, as well as the optic tracts and olfactory nerves.
These findings contribute to the scarce body of literature pertaining to the neuroimaging manifestations of MPV17-related mitochondrial DNA depletion syndrome, despite the illness being largely defined by its neurological presentation. Since no specific patterns have been identified which would enable imaging to be used as a reliable diagnostic tool, these results establish a robust foundation to promote further exploration in this field [6, 7, 10, 16].
In cases where the perirolandic cortices in neonates and hippocampi in older infants were affected, there was no history of asphyxia, signal changes were acute, as evidenced by restricted diffusion, and there were no accompanying basal ganglia or thalamic changes to suggest hypoxic ischemic injury.
For those patients with genetically confirmed MPV17-related mitochondrial DNA depletion syndrome in whom initial neuroimaging is normal, follow-up MRI would enhance our understanding of the natural history of the condition. However, it may not always be feasible due to the rapidly progressive trajectory of this form of the disease, subjecting patients to early demise (mean age of death approximately 10 months) or the development of complications that preclude general anaesthetic. None of the patients in our cohort underwent follow-up neuroimaging.
An additional consideration is that most patients with MPV17-related mitochondrial DNA depletion syndrome present with cholestatic jaundice. It follows that hyperintense signal in the globus pallidi, identified in five of eight (62.5%) of the MRI studies, may indicate concurrent bilirubin encephalopathy, particularly in the two patients where the abnormality was linear and confined to the posteromedial aspects. The full diagnostic imaging triad of injury to the CA1 and CA2 sectors of the hippocampi and subthalamic nuclei was not, however, observed [17].
Furthermore, since the administration of contrast did not result in pathologic enhancement in any cases, it is probably not indicated when strong clinico-radiological evidence exists for MPV17-related mitochondrial DNA depletion syndrome.
This study has two primary limitations. Firstly, we did not compare our findings to other mitochondrial disorders or different variants of MPV17-related mitochondrial DNA depletion syndrome, which prevents us from determining the specificity of our results to MPV17-related mitochondrial DNA depletion syndrome. Secondly, despite being the largest single-centre, single-variant cohort in the literature, our patient population is still relatively small, limiting the statistical significance of our findings. In contrast, the literature on better-known mitochondrial disorders like Leigh syndrome has identified characteristic imaging manifestations. Spongiform lesions of the basal ganglia (particularly the putamina) and brainstem, with a predilection for the midbrain and periaqueductal grey matter, are commonly observed. A study published in AJNR in 2000 analysed the MRI findings of eight patients with known Leigh syndrome and found that half had lesions conforming to the lower brainstem, reticular formation, or reticulospinal tracts. The authors proposed that the detection of lower brainstem lesions at the onset of respiratory abnormalities might be specific for Leigh syndrome [18]. We now know that this is not the case and that such changes may be a manifestation of a broader range of mitochondrial conditions.

Comments (0)