
Remember me
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by a general breakdown of immune tolerance leading to the production of pathogenic autoantibodies, autoreactive immune cells and proinflammatory cytokines [1]. Abnormal effectors of the immune system act in concert with distinct pathways expressed by tissue-resident cells to generate inflammation and organ failure [2]. In recent years, studies that explore the metabolic pathways that shape immune cell function have revealed that the expression of distinct immune cell subsets and the aberrant production of cytokines, including decreased production of interleukin (IL)-2 and IL-17, are governed by intracellular metabolic processes. Research on these processes has revealed a number of metabolites that can control the autoimmune response and subsequent organ inflammation [3]. Key metabolic pathways, including glycolysis, oxidative phosphorylation, the tricarboxylic acid cycle, and the pentose phosphate pathway, are known to be disrupted in SLE T cells, contributing to their abnormal activation and differentiation [4]. A study published in Cellular & Molecular Immunology [5] highlighted a previously underappreciated metabolic contributor to the pathogenesis of SLE, the farnesoid X receptor (FXR), a nuclear receptor primarily known for its role in bile acid homeostasis and cholesterol metabolism. This study revealed that FXR plays a critical immunoregulatory role by promoting the expansion of T follicular helper (Tfh) cells in lupus. Treatment of lupus-prone mice with the FXR signaling inhibitor ursodeoxycholic acid (UDCA) mitigated disease progression, suggesting that it represents a potential therapeutic target for patients with SLE (Fig. 1).
Fig. 1
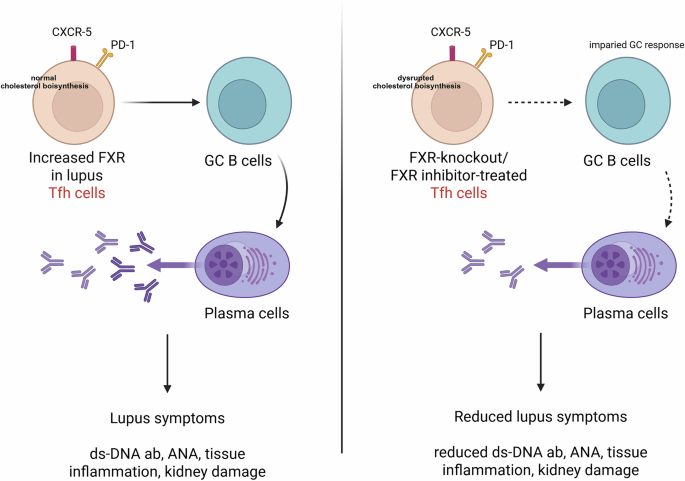
FXR promotes T follicular helper (Tfh) cell differentiation in mouse models of SLE. Left, Increased FXR expression in lupus Tfh cells. Tfh cells help germinal center (GC) B cells generate plasma cells and produce antibodies that contribute to lupus pathology. Right, FXR deficiency or treatment with an FXR inhibitor leads to impaired GC responses and fewer plasma cells. Abbreviations: ds-DNA, double-strand DNA; ab, antibody; ANA, antinuclear antibody
Wang et al. [5] first studied the gene and protein expression of FXR in the main subsets of immune cells in the peripheral blood of SLE patients. They reported increased FXR expression in Tfh cells from the peripheral blood of SLE patients compared with other immune subsets, suggesting a potentially unique role for FXR in T-cell-driven autoimmunity. Tfh cells are a subset of CD4⁺ T cells crucial for the formation of germinal centers (GCs) and the production of high-affinity antibodies by B cells. In subsequent mechanistic experiments, Wang et al. studied the effect of FXR on Tfh cells. To determine the functional consequences of FXR in Tfh biology, the authors used FXR knockout (Fxr−/−) mice. These mice presented a marked reduction in both Tfh cell numbers and GC formation. This finding was further substantiated in a pristane-induced lupus model, where compared with control mice, FXR-deficient mice presented significantly attenuated lupus pathology, including smaller spleens, decreased proteinuria, and reduced autoantibody levels. These results demonstrated that FXR contributes to lupus pathogenesis through the promotion of aberrant Tfh responses. The next question the authors addressed was how FXR regulates Tfh cells. They performed RNA sequencing of Tfh cells isolated from wild-type and Fxr−/− mice. Transcriptomic analysis revealed that FXR regulates genes involved in cholesterol biosynthesis and homeostasis, which in turn influences Tfh cell proliferation and survival. This link between lipid metabolism and immune cell function underscores the concept of broad crosstalk between metabolic processes and autoimmunity. Finally, to assess the translational relevance of these findings, they tested the therapeutic potential of targeting FXR signaling in vivo. The bile acid derivative UDCA, a clinically approved drug known to inhibit FXR signaling, was administered orally to mice in which lupus was induced by pristane and to MRL.lpr mice, which develop lupus spontaneously. In both settings, treatment led to a significant reduction in disease severity, characterized by fewer Tfh and GC B cells, lower serum levels of autoantibodies, and overall improved clinical outcomes. These promising results point to FXR as an important therapeutic target in SLE and highlight the potential of repurposing FXR-modulating drugs, such as UDCA, to treat autoimmune diseases.
Although the rapid translation of basic science findings to clinically relevant therapeutic tools is exciting, it is essential to acknowledge its limitations and questions that were not addressed by this study. First, while the FXR inhibitor UDCA has demonstrated therapeutic efficacy in pristane-induced and MRL/lpr lupus models, it has previously failed to significantly benefit NZB × NZW F1 lupus-prone mice [6]. This discrepancy raises important concerns about the influence of pathogenetic heterogeneity, which is prevalent in patients with SLE and suggests that UDCA may offer clinical benefit only in a fraction of patients. Second, the current findings are based on murine models, which, although they provide valuable mechanistic insights, cannot predict whether inhibition of FXR in patients with lupus will have any clinical benefit. Therefore, further studies using ex vivo approaches or lupus-humanized mice are needed prior to planning any clinical trials. It remains to be determined why FXR is upregulated in Tfh cells. It is possible that its upregulation represents previously known biochemical and molecular pathways [7] operating in multiple immune cells or aberrant pathways limited to the expression of FXR in Tfh cells. Along these lines, it is important to know whether global targeting of FXR affects the function of other immune cells. Addressing these questions will be critical to fully understand the role of FXR in SLE and to evaluate its value as a therapeutic target.
Cholesterol and lipid metabolism are increasingly recognized as key contributors to the pathogenesis and progression of SLE. Patients with SLE frequently present with dyslipidemia, including elevated low-density lipoprotein, decreased high-density lipoprotein, and increased triglycerides, all of which promote accelerated atherosclerosis, a leading cause of mortality in patients with lupus. Statins, which inhibit cholesterol synthesis, are frequently used to manage hyperlipidemia and reduce cardiovascular risk in patients with SLE. However, while generally well tolerated, statins cause side effects, including muscle and joint pain, nausea, and constipation, along with more severe adverse effects, such as liver toxicity and severe muscle damage. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a key regulator of cholesterol metabolism, and elevated PCSK9 levels in SLE patients are correlated with increased disease activity and systemic inflammation. As such, PCSK9 inhibitors (e.g., evolocumab and alirocumab) represent promising alternatives for SLE patients who are intolerant to statins or fail to achieve adequate lipid control [8]. Additionally, apolipoprotein A-I mimetic peptides, which promote cholesterol efflux and exhibit anti-inflammatory properties, have shown potential in preclinical models of lupus. These peptides reduce systemic inflammation and ameliorate atherosclerosis-related complications [9]. However, additional investigations are needed to evaluate their safety, toxicity, and efficacy in human clinical trials before they can be considered viable therapeutic agents for atherosclerosis in SLE patients.
Comments (0)