
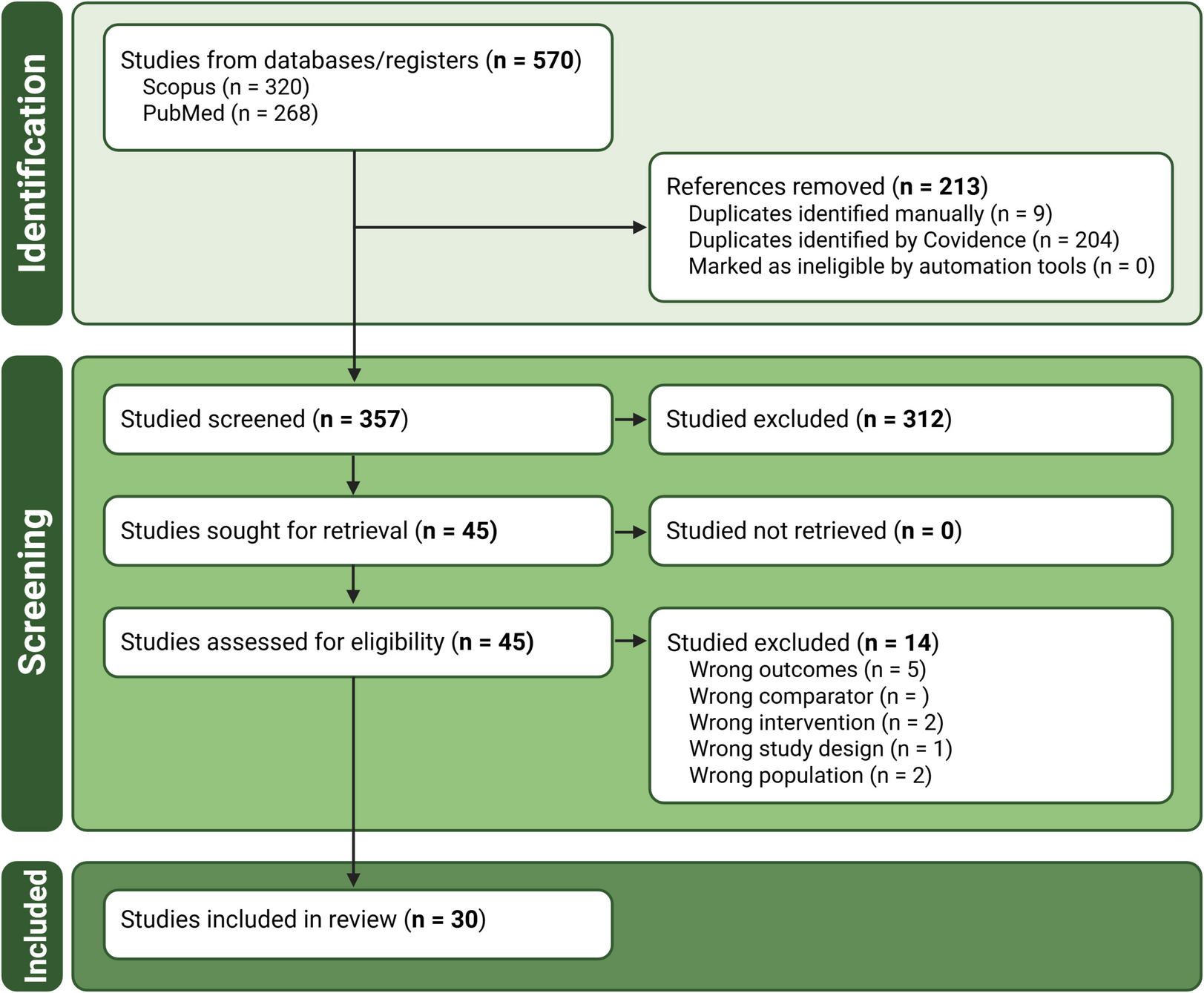
Remember me
SD is implicated in several neurological and neurovascular disorders [6,7,8,9], which are often characterised by neuroinflammation [10,11,12]. Investigating a potential bidirectional relationship between SD and inflammation may yield new therapeutic targets for neurological disorders which are worsened by SD. We therefore set out to investigate if SD induces neuroinflammatory effects and if increased inflammatory states results in exacerbated SD responses.
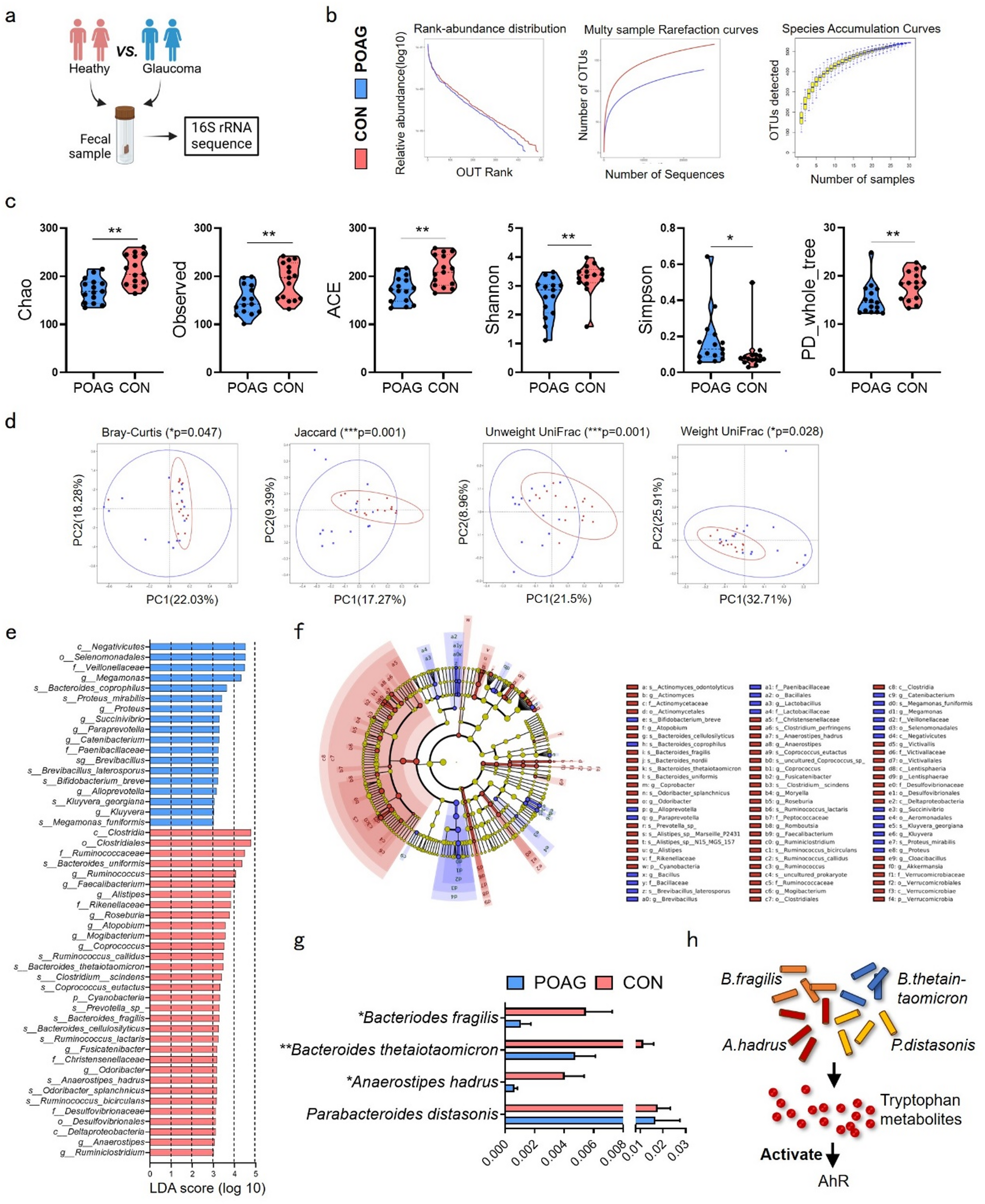
The majority of studies found an upregulation in inflammatory markers following SD, including IL-1β [13, 16, 17, 22,23,24,25,26, 29, 30], TNF-α [13, 16, 22,23,24, 30, 31], IL-6 [13, 16, 25] and COX-2 [16, 17, 25,26,27, 30, 32, 33] (Fig. 4). This was accompanied by evidence of neuroinflammatory cell activation, including reactive astrogliosis and microglial morphological changes [14, 27, 30, 37, 38]. Several studies also reported an increase in extracellular HMGB1 release [17, 30, 34,35,36] and NF-κB signalling [17, 25, 34], indicating a sustained neuroinflammatory response. Additionally, upregulation of CGRP [24, 26, 30, 33] was also reported, suggesting a neurogenic inflammatory component.
Fig. 4
Overview of the results of the systematic review. Spreading depolarization (SD), induced via electrical, pinprick, optogenetic, or potassium chloride (KCl) stimulation, triggered upregulation of inflammatory mediators and cellular responses. In models of altered inflammation, including brain injury, peripheral inflammation, and encephalomyelitis, only TNF administration reduced SD amplitude
Three studies examined the effects of altered inflammatory states on SD characteristics, yielding mixed evidence, likely due to the diversity of models used [18, 43, 44], with one study reporting a reduction in SD amplitude following inflammation [18].
The effect of methodological variability in SD induction on inflammatory profilesThere was wide variation in the methods used to induce SD. While most studies used KCl to induce SD, there were several studies which induced SD via pinprick for at least one subgroup [17, 24, 26, 34, 36, 40]. SD induction often involves craniotomy, which has been shown to confound the inflammatory response [45]. Therefore, cortical injuries, caused by either pinprick or invasive procedures, may have influenced study findings. Three studies comparing optogenetic-induced SD with invasive methods found no differences in inflammatory markers, suggesting that the method of SD induction did not influence the outcomes [16, 30, 36].
SD elicits a sustained neuroinflammatory responseInflammatory markers were assessed in various samples, including ex vivo brain slices [13, 23], cultured astrocytes [13], cortical tissue [14, 16, 17, 22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39], CSF-enriched solutions [30], trigeminal ganglion [30], dura mater [40], and pial microvessels [41]. Interestingly, while significant changes in inflammatory markers were observed in cortical tissue compared to sham samples, no differences were detected in plasma. This suggests that the inflammatory changes are localized specifically to the neuronal tissue [46].
IL-1β was the most studied inflammatory cytokine in this review and was mostly found to be upregulated following SD [13, 16, 17, 22,23,24,25,26, 29, 30]. In a study by Takizawa, T et al., IL-1β expression was significantly increased as soon as 10 min following SD and peaked at 1 h, supporting the notion that IL-1β is one of the first inflammatory cytokines released following SD [16]. A further study suggested a possible mechanism for this increase, determining that the increase in IL-1β release following SD was dependent on the release of the NLR family pyrin domain containing 3 (NLRP3) inflammasome [30]. Knockout of the IL-1β-encoding gene in mice also ameliorated the increase in COX-2 post-SD [30]. This may suggest IL-1β may partially mediate downstream signalling following SD, with NLRP3 as the upstream mediator of this inflammatory axis.
Other cytokines which were commonly found to be raised following SD in the cortical tissue, include TNF-α [13, 16, 22,23,24, 30, 31] and IL-6 [13, 16, 25]. Knockout of the gene encoding interleukin-1 receptor (IL-1R) gene resulted in a blunting of the TNF-α response following SD in one study [16]. Another study found that administration of TLR2/4 was able to suppress microglial activation following SD without reducing IL-1β levels yet was still able to cause a reduction in cortical TNF-α mRNA expression [30]. These findings may suggest that the rise in TNF-α following SD occurs due to a combination of upstream IL-1β action and microglial modulation. IL-6 exhibits both pro- and anti-inflammatory properties and contributes to the induction of acute-phase responses [47]. It plays a key anti-inflammatory role in local and systemic acute inflammation by modulating proinflammatory cytokine levels without affecting anti-inflammatory cytokines [47]. The mixed findings following SD [13,
Comments (0)