In this study, the girl was diagnosed with PD through a combination of clinical manifestation, genetic testing, and functional experiments. She presented with ILD as the predominant manifestation, accompanied by facial deformities, short stature, developmental delay, hypothyroidism, anemia, and hypergammaglobulinemia (elevated levels of IgG and IgE). Using WES and Sanger sequencing identified a novel homozygous variant of the PEPD gene (c.741–13 C > A), leading to abnormal pre-mRNA splicing, which was confirmed by a minigene assay. Specifically, two abnormal transcripts, r.741-1_741-11ins and r.741-1_741-51ins, were transcribed by a minigene plasmid expressing the c.741–13 C > A variant, thereby confirming the pathogenicity of the variant.
Since the first report by Goodman.et al. in 1968 [1], approximately 200 cases with PD have been documented worldwide [3,4,5,6,7]. The global prevalence of PD remains unknown. However, certain populations exhibit elevated carrier frequencis due to founder variants, notably among the Ohio Amish in the USA [8], as well as the Druze and Arab Muslims in Northern Israel [9]. PD is exceptionally rare among the Chinese population. Since 1989, only three sporadic cases have been reported, with genetic results available for only one of these cases [3, 10, 11].
The clinical phenotype of PD exhibits significant heterogeneity, ranging from asymptomatic cases to potentially life-threatening conditions such as respiratory insufficiency, severe hepatitis, and cardiorenal amyloidosis [6]. Notably, Rossignol et al. reported that the most frequently manifestations are dermatologic lesions (84%), followed by dysmorphic features (67%), developmental anomalies (58%), recurrent or severe infections (48%), and splenomegaly (45%) between 1968 and 2020 [6]. Dermatological lesions may not present as the first signs of PD; instead, they are part of a spectrum of symptoms that emerge gradually from the neonatal stage through to adulthood [12].
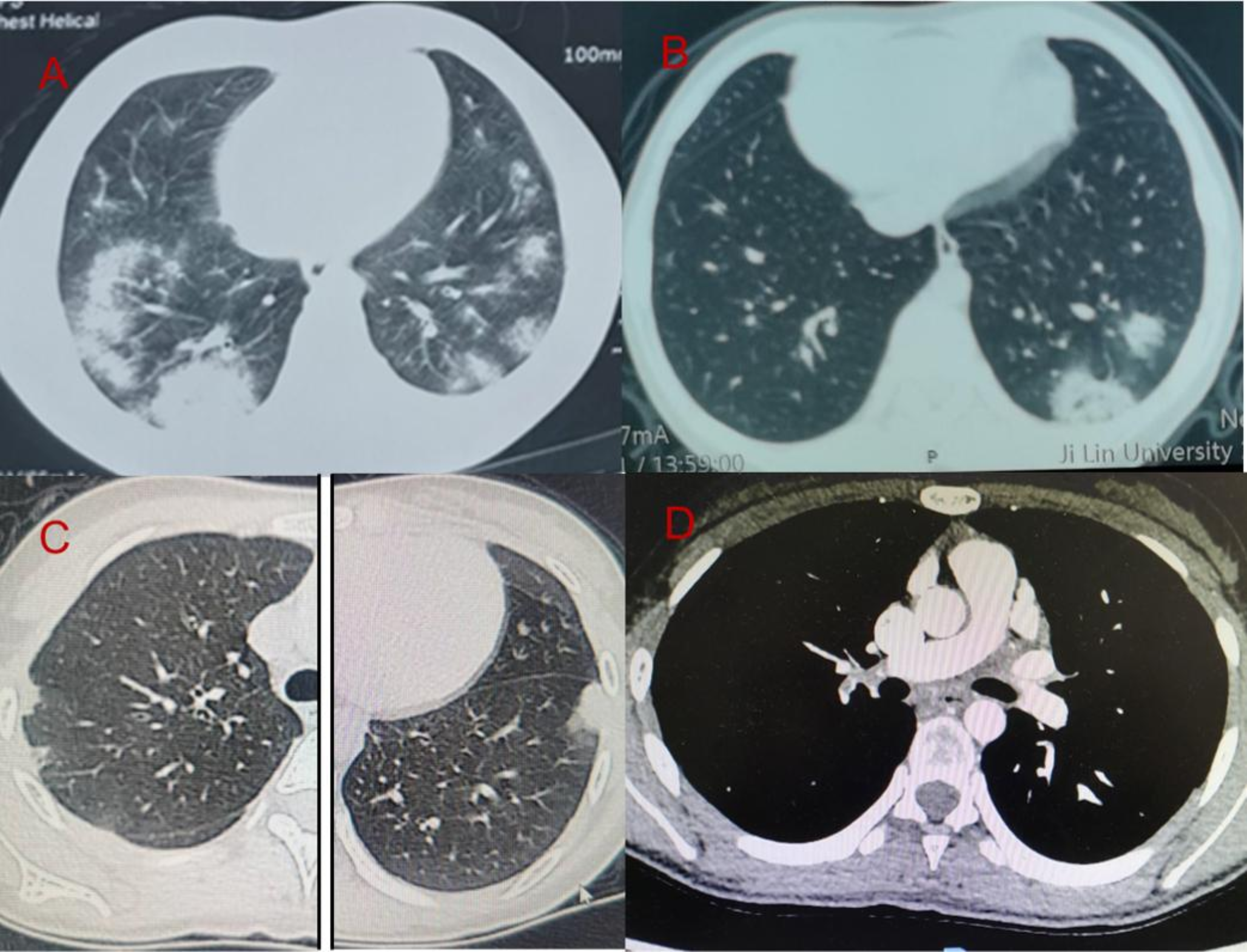
Pulmonary involvement in PD occurs in approximately 26% of cases, primarily manifesting as recurrent or severe respiratory infections and asthma [3,4,5,6,7]. ILD is the rarest pulmonary presentation, with only eleven reported cases globally, including five in Israel [13, 14] and one each in the USA [4], Portugal [15],China [3], Japan [16], and South Asia [17], with varying degrees of severity. Chest imaging findings are varied and may present as cystic changes, ground-glass opacities, bronchiectasis, reticulation, thickening along septal and peribronchovascular lines, emphysematous changes, and air trapping. While the exact mechanisms of ILD in PD remain elusive, it is associated with disrupted collagen recycling, cell death resembling necrosis, and heightened oxidative stress, which may lead to the gradual deterioration of lung tissue [13]. Cases primarily presenting with ILD but without dermatologic lesions are exceedingly rare in PD. This highlights the importance of considering PD in the differential diagnosis of ILD, regardless of the presence of dermatologic lesions. This also emphasizes the extensive clinical spectrum of PD and the potential for misdiagnosis or underdiagnosis. Given the patient’s youth, the emergence of typical dermatological lesions warrants continued observation.
Certain PD patients exhibit mutations in the PEPD gene, located at 19q13.11. According to the Human Gene Mutation Database (July 2023), there are 61 documented PEPD mutations, including 38 missense/nonsense, 7 splice site, 10 microdeletions, 2 microduplications, and 3 large deletions. Notably, a prevalent mutation, c.605 C > T, accounts for 85% (17/20) of PD cases in Israel [14]. However, the genotype-phenotype relationship remains unclear. Rossignol et al. indicate that biallelic missense variant carriers tend to develop ulcers less frequently and later than those with loss-of-function variants [6]. In our case, a child with a homozygous mutation inherited from both parents suggests an ancestral link, despite no reported consanguinity. Additionally, a novel splice site mutation, c.741–13 C > A, disrupts pre-mRNA splicing, resulting in two abnormal transcripts: r.741-1_741-11ins and r.741-1_741-51ins, as confirmed by a minigene assay, thus establishing its pathogenicity. This discovery expands the spectrum of known PEPD gene mutations.
The treatment of PD remains symptomatic, with no recommended or curative regimen. For managing cutaneous ulcers, topical and systemic treatments only provide partial temporary benefit. For autoimmunity, rituximab has shown promising efficacy [18]. Efforts to replace prolidase activity, including blood transfusions, gene therapy, and enzyme replacement, have shown limited efficacy [12]. The efficacy of hematopoietic stem cell transplantation remains controversial [19, 20]. Hyperimmunoglobulinemia E is common in PD due to the binding of gamma globulins to the prolidase substrate in serum and as a result of repeated infections and immunological dysregulation [2]. The efficacy of corticosteroids on cutaneous lesions is likely mediated by inhibiting the infiltration of polymorphonuclear leukocytes and the generation of superoxide by neutrophils [21]. However, the role of corticosteroids in patients with ILD remains unassessed. A long-term follow-up study by Cottin et al. indicated that while fibrosis may have become less pronounced over time, emphysema and cystic changes continued to progress, ultimately leading to chronic respiratory insufficiency [15]. Interestingly, our patient’s response to corticosteroid therapy diverged from Cottin et al.‘s findings. After 5 months of treatment, our patient demonstrated significant improvement in exercise capacity and a gradual decline in KL-6 levels, which was consistent with radiological recovery. Additionally, the notable reduction in respiratory infections at the 1-year follow-up indicates a satisfactory therapeutic outcome.



Comments (0)