
Remember me


Patient 1 (P1), born to non-consanguineous parents, initially presented with recurrent oral ulcers and intermittent fevers at 6 years and 7 months old, poorly responding to antimicrobial therapy. Ulcers appeared on the tonsils, tongue, lips, and buccal mucosa, occasionally with Candida albicans infection. At 7 years old, she began experiencing recurrent abdominal pain, moderate anemia, and was underweight (-2SD). Her mother had recurrent oral ulcers with no treatment and further disease progression. Her brother died at 6 years old after recurrent fever, oral ulcers, and bloody stools. She has 2 healthy younger sisters. Immunological tests were normal. At 7 years and 1 month old, colonoscopy revealed ulcers in the terminal ileum and colon, leading to a diagnosis of Crohn’s disease. She was treated with glucocorticoids, thalidomide, and infliximab, showing sustained remission. After more than 20 doses of infliximab, unexplained abdominal pain prompted switching to vedolizumab, which eased her pain. She continues maintenance treatment with vedolizumab and oral ulcers did not recur.
Patient 2 (P2), a 7-year-old girl, began showing symptoms at 3 years of age, with recurrent oral, vulvar, and perianal ulcers. Despite symptomatic treatment, she frequently relapsed and also experienced recurrent fevers and erythema nodosum-like rashes. Elevated CRP (C-reactive protein), ESR (erythrocyte sedimentation rate), and transient hip synovitis were noted. Her uncle had recurrent oral ulcers, and her maternal grandmother was suspected of having Sjögren’s syndrome. Immunological tests showed normal Ig (Immunoglobulin) levels and negative autoantibodies, but elevated anti-β2-glycoprotein I antibodies and lupus anticoagulant. Colonoscopy revealed a terminal ileum ulcer and rectal erosions. She was treated with mesalamine, steroids, and adalimumab, achieving sustained remission.
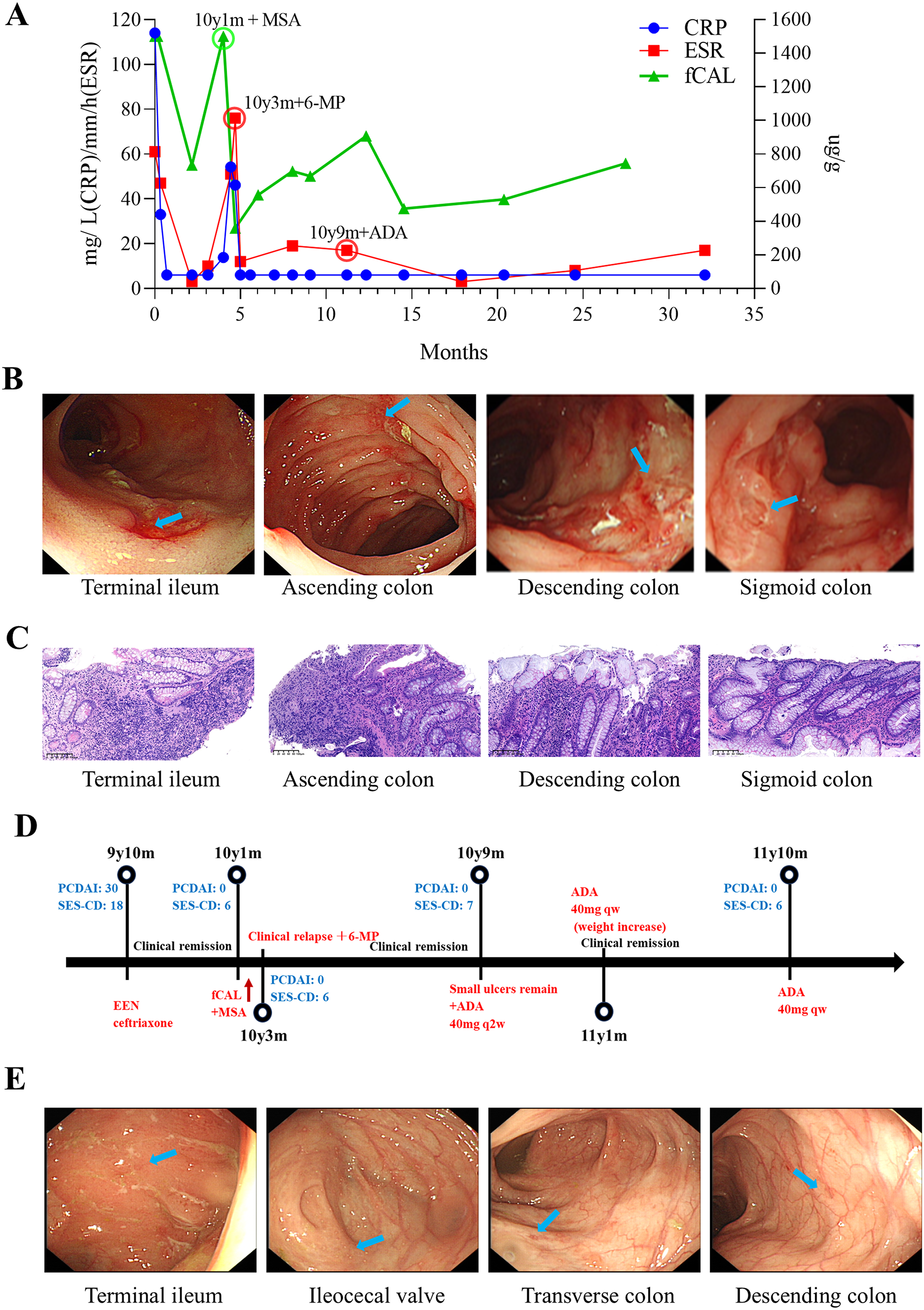
Patient 3 (P3), a 12-year-old girl, started showing symptoms at 9, including recurrent oral ulcers, diarrhea, weight loss, and fever, with elevated CRP and ESR (Fig. 1A) and poor response to antimicrobials alone. Her father was suspected of having irritable bowel syndrome (IBS). Stool tests revealed pyocytes. Immunological tests showed elevated T and B cells, and increased TNF-alpha, IL-1beta, and IL-6 in the serum. Fecal calprotectin was over 1500ug/g (<50 ug/g) (Fig. 1A). Colonoscopy revealed deep ulcers from the cecum to the sigmoid colon and pathological biopsy of the corresponding sites revealed inflammatory changes with ulcer formation (Fig. 1B and C). Diagnosed with Crohn’s disease, P3 was treated with EEN (Exclusive enteral nutrition), and later mesalamine, 6-mercaptopurine, and finally adalimumab, achieving sustained remission (Fig. 1D and E, Supplemental Figs. 1–2).
Fig. 1
Clinical course of patient 3. A. Time line of C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and fecal calprotectin (fCAL) levels over 2 years. B. Representative colonoscopy images at the age of 9 years and 10 months old (Scattered ulcers were observed in the terminal ileum; from the cecum to the sigmoid colon, there were scattered deep large ulcers arranged in a longitudinal pattern, particularly notable in the transverse colon, descending colon, and sigmoid colon, with normal mucosa between the lesions) and corresponding H&E micrographs C. Representative histopathological H&E staining at the age of 9 years and 10 months old (terminal ileum: No epithelium observed. The lamina propria showed infiltration by numerous lymphocytes, plasma cells, scattered medium and long granulocytes, and eosinophils. Ascending colon: Diffuse neutrophils, lymphocytes, plasma cells, and scattered eosinophils were present in the lamina propria. Inflammation caused epithelial and glandular destruction, leading to ulcer formation. Descending colon: Diffuse neutrophils, abundant lymphocytes, plasma cells, and scattered eosinophils were observed in the lamina propria. Crypt abscesses were present, with inflammation destroying the epithelium and glands, resulting in ulcers. Sigmoid colon: Diffuse neutrophils, numerous lymphocytes, plasma cells, and scattered eosinophils were found in the lamina propria. Inflammation led to epithelial and glandular destruction, forming ulcers). Scale bar, 100 μm. D. Timeline of colonoscopy inflammation scores and treatment course. E. Representative colonoscopy images at the age of 10 years and 9 months old indicating inflammation relapse (Scattered superficial ulcers with white exudate on the surface were observed in the terminal ileum, scattered aphthous ulcers in the cecum, minor erosions in the transverse colon, and linear white scars in the descending colon. The remaining intestinal mucosa appeared normal). MSA: Mesalazine, 6-MP: Mercaptopurine, ADA: adalimumab. PCDAI: Pediatric Crohn’s Disease Activity Index, SES-CD: Simple Endoscopic Score. Blue arrow pointing at typical lesion sites
A brief summary of clinical and genetic findings of these patients was presented in Table 1.
Detailed clinical information about these patients can be found in Supplementary data.
Table 1 Clinical manifestations summary of novel female DEX patientsGenetic FindingsDue to the young age of onset or positive family histories, whole-exome sequencing was performed for the parents and the affected children. P1, her mother who has recurrent oral ulcers, and her healthy second younger sister are all heterozygous carriers of the c.320(exon4)_c.321(exon4)insA or p.L108fs*3 variant in ELF4, but not her father (Fig. 2A). P2 is heterozygous carrier of the c.329(exon4)delA or p.E110Gfs*35 variant in ELF4, but not her parents, indicating a De Novo mutation (Fig. 2B). However, her uncle and maternal grandmother have a history of oral ulcers, with unknown genotypes. P3 is heterozygous carrier of the c.685(exon7)A > G or p.I229V variant in ELF4 and her father is hemizygous of this variant (Fig. 2C). All these variants were confirmed by Sanger sequencing.
Fig. 2
Three families carrying the ELF4 mutation. A. Pedigree of family 1 (left) and Sanger sequence reads demonstrating locations of the variants: WT, wild type; Mut, c.320_c.321insA variant; grey circle, asymptomatic carrier. B. Pedigree of family 2 (left) and Sanger sequence reads demonstrating locations of the variants: WT, wild type; Mut, c.329 delA variant. C. Pedigree of family 3 (left) and Sanger sequence reads demonstrating locations of the variants: WT, wild type; Mut, c.685 A > G variant. X, X chromosome; Y, Y chromosome
Pathogenicity Analysis of the VariantsTheoretically, the frameshift mutations in P1 and P2 would lead to the production of a truncated ELF4 protein of over 100 amino acids in length. We constructed mutant expression plasmids and found no wild-type ELF4 protein but rather the truncated ELF4 protein was detected (Fig. 3A). Additionally, using the previously reported IFN-b dual-luciferase reporter assay, we showed the truncated ELF4 proteins failed to activate the IFN-b promoter, likely due to the failure to express important domains such as the NLS and ETS domains (Fig. 3B). The I229V missense mutation carried by P3 is located in the ETS domain. This variant does not occur in the 1000 Genomes database, ExAC database, or in-house database. This amino acid site is highly conserved across species and among ETS domain transcription factor superfamily members [3]. Pathogenicity prediction using multiple methods classified this variant as “Damaging” (Supplementary Table 1). The AlphaMissense pathogenicity score of this variant is 0.948 (likely pathogenic) [13]. Structurally, various atomic contacts and bonds are predicted to be disrupted by the mutation [14, 15] (Fig. 3C). Similarly, the I229V ELF4 failed to activate the IFN-b promoter (Fig. 3B), although it has little impact on protein expression levels (Fig. 3A, D and E).
Fig. 3
Pathogenicity evaluation for novel variants of ELF4. A. Plasmids constructs mimicking patients’ variants were expressed in HEK293T cells and determined by western blot. B. Quantification IFN-β dual luciferase reporter activity in HEK293T cells transfected with empty vector (EV), WT and mutant ELF4 variants. Data were pooled from 3 independent experiments and represented as mean ± SD. ****P < 0.0001. Unpaired two-sided Student’s t test. C. Interatomic interaction changes predicted by DynaMut using AlphaFold2-predicted ELF4 structure. Wild-type and mutant residues are colored in light-green and are also represented as sticks alongside with the surrounding residues which are involved on any type of interactions. D. The expression of ELF4 variants in P3 was determined by western blot using PBMCs. Representative images from 2 independent experiments. (E) Cycloheximide chase assay for I229V mutant ELF4 and WT control. Representative images from 2 to 3 independent experiments. (F) Schematic diagram of ELF4 protein and the position of ELF4 variants (upper: from this paper, lower: from previous reports). W231R mutant ELF4 was used as positive control [3]
X Chromosome Inactivation AnalysisAccording to previous reports, DEX tends to follow an X-linked recessive inheritance pattern, where female carriers typically do not develop symptoms [16]. Skewed X-chromosome inactivation is one of the main mechanisms causing female carriers to develop symptoms in X-linked recessive diseases [17, 18]. Therefore, we examined the X-chromosome inactivation in the three probands. We found that P1’s paternal X-chromosome (carrying the wild-type ELF4 genotype) had an inactivation rate of approximately 82%, indicating moderate skewing of inactivation (> 70%, < 90%) (Fig. 4A), leading to overexpression of the mutant ELF4. In contrast, her second younger sister, who also carries the mutation but does not exhibit symptoms, did not show X-chromosome inactivation skewing (53% <70%) (Fig. 4A). P3’s maternal X-chromosome (carrying the wild-type ELF4 genotype) had an inactivation rate of about 83%, also indicating moderate skewing of inactivation (Fig. 4B). The polymorphic locus of the AR gene cannot distinguish between the paternal and maternal X chromosomes of P2. Therefore, we employed another method (with additional 3 polymorphic genetic markers), which revealed that P2’s paternal X-chromosome had an inactivation rate of approximately 33.7% identified as skewed in this system (≥ 25%) (Fig. 4C). Since the ELF4 mutation carried by the proband is De novo, we cannot determine whether it occurred on the maternal or paternal X-chromosome.
Fig. 4
Skewed X chromosome inactivation patterns in affected female DEX patients. A. XCI pattern in PBMCs from members of family 1 using traditional method (the HUMARA locus). Representative result from 2 independent experiments. B. XCI pattern in PBMCs from members of family 3 using traditional method (the HUMARA locus). Representative result from 2 independent experiments. C. XCI pattern in PBMCs from members of family 2 using optimized method (additional locus). This result is from one single experiment
Comments (0)