
Remember me


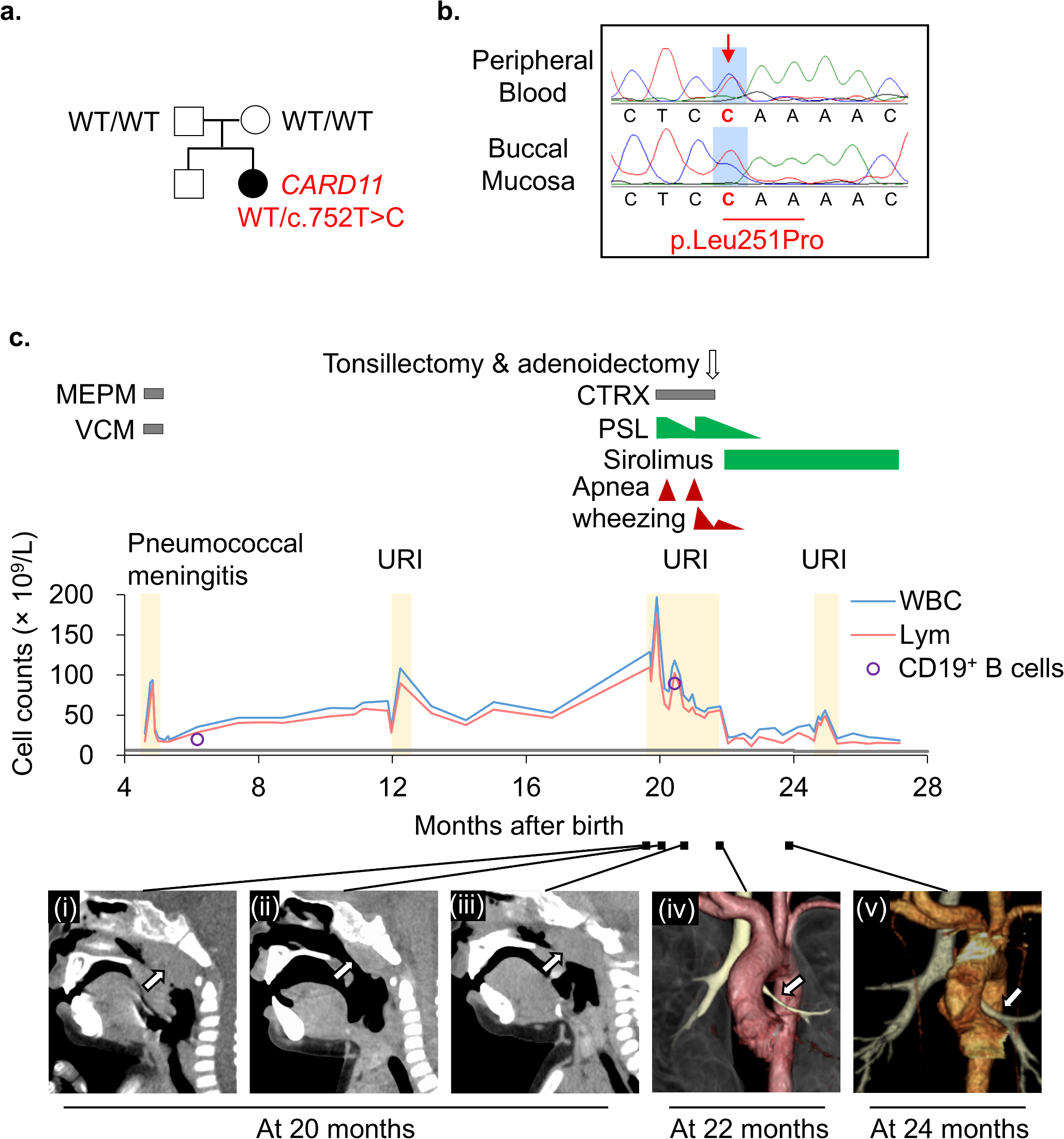
A 5-month-old infant presented to the hospital due to leukocytosis. The infant was delivered vaginally at 39 3/7 weeks gestation, weighing 3,030 g. At 4 months of age, the patient experienced bacterial meningitis caused by Streptococcus pneumoniae serotype 35B, which is not covered by the 13-valent pneumococcal conjugate vaccine. The patient received appropriate antimicrobial treatment and did not exhibit any apparent neurological complications. However, the patient was referred to the hospital due to the persistent leukocytosis of 35.5 × 109/L. The patient did not have a family history of immunodeficiency diseases or genetic disorders. During the physical examination, mild splenomegaly was observed, but no palpable lymphadenopathy was detected. Blood tests revealed an elevated white blood cell (WBC) count of 35.5 × 109/L, primarily consisting of B-lymphocytes. Additionally, mild anemia was detected with a hemoglobin level of 104 g/L, while the platelet count was 308 × 109/L (Table S1). The serum IgG level was found to be elevated at 16.43 g/L. Flow cytometric analysis revealed an increase in CD19+ B cells, particularly in the naïve B-cell subset (CD19+CD20+CD27−IgD+), without an elevation in double-negative T cells (CD3+TCR-αβ+CD4−CD8−) (Table S1). Southern blotting of the IGH gene rearrangement demonstrated a polyclonal B-cell lymphocyte proliferation pattern (Fig. S1a). Analysis of the bone marrow aspirate revealed normal trilineage blood cell differentiation with no presence of immature blasts but an increased count of normal lymphocytes (Fig. S1b). WES identified a heterozygous missense variant in the CARD11 gene (NM_032415, c.752T > C, p.Leu251Pro; Fig. 1a,b and S2a). Sanger sequencing of proband and family samples confirmed the identified variant as a de novo germline variant. Based on this finding, the patient was diagnosed with BENTA, characterized by a gain-of-function germline mutation in the CARD11 gene. Without treatment, the patient’s polyclonal B-cell lymphocytosis continued to worsen. As a result, at 20 months of age, the patient was hospitalized for an upper respiratory tract infection and wheezing, which was attributed to marked adenoid hypertrophy (Fig. 1c). The patient’s WBC count was markedly elevated at 196.9 × 109/L, and she experienced nasal congestion and apnea during sleep, necessitating the use of oxygenation. To address these symptoms, prednisolone (1.5 mg/kg/day) was administered, resulting in the shrinkage of enlarged tonsils and a decrease in the WBC count. However, as the prednisolone dosage was tapered, the patient’s respiratory symptoms worsened once again. As a result, she underwent an adenoidectomy and right tonsillectomy, which successfully resolved the wheezing. Unfortunately, 1 month later, the wheezing recurred, and the lymphocyte count increased once more. A chest computed tomography (CT) scan revealed bronchial stenosis and mediastinal lymphadenopathy caused by peribronchial tissue proliferation. The patient’s treatment was modified to include sirolimus at a dosage of 1 mg/m2/day, along with the gradual withdrawal of prednisolone. After 2 months, the patient’s wheezing completely disappeared, and subsequent CT scans revealed improvement in the bronchial stenosis and peribronchial tissue proliferation, with only minimal changes observed in the mediastinal lymph nodes and spleen. Thereafter, it is worth noting that the patient’s respiratory status remained stable with sirolimus. At 24 months of age, the patient encountered an upper respiratory tract infection and transient leukocytosis, with a maximum WBC count of 55.5 × 109/L, which improved spontaneously without the development of bronchial stenosis. Over the subsequent months, the spleen size gradually decreased, and by 10 months after the initiation of sirolimus, the splenomegaly was no longer palpable. The patient was maintained on sirolimus monotherapy.
Fig. 1
Patient characterization. (a) The family pedigree. A heterozygous c.752T > C, p.Leu251Pro variant was identified by whole-exome sequencing. The variant was not present in the unaffected parents. (b) Sanger sequencing of the CARD11 variant in the proband. (c) Presentation of the clinical course. The patient had persistently elevated leukocytic and lymphocyte counts, which increased sharply following an upper respiratory tract infection, resulting in apneic attacks and wheezing. The gray line at the lower end of the graph indicates the age-related upper limits for absolute lymphocyte counts. Administration of prednisolone (PSL), followed by tonsillectomy and adenoidectomy, improved the apneic attacks. Subsequent treatment with sirolimus led to further reductions in white blood cell count and improvements in wheezing. (i) Computed tomography (CT) scans revealing upper airway stenosis caused by adenoid hypertrophy triggered by respiratory infection. (ii) Temporary shrinkage was observed with PSL treatment. (iii) However, the condition worsened again after PSL tapering. (iv) Three-dimensional CT imaging of the tracheobronchial tree. The left bronchus (arrow) shows stenosis due to peribronchial tissue proliferation. (v) This stenosis improved after sirolimus treatment. WT, wild-type; MEPM, meropenem; VCM, vancomycin; CTRX, ceftriaxone; PSL, prednisolone; URI, upper respiratory tract infection; WBCs, white blood cells; Lym, lymphocytes
Literature ReviewDuring the literature search, 229 records were initially identified. Out of these, 52 reports were selected for further screening based on their titles and abstracts. Additionally, 50 reports were identified for retrieval to examine their full texts. After the screening and retrieval process, 15 eligible articles were selected, and a manual literature search identified one additional article. Thus, a total of 16 eligible articles in English were included, which provided information on 34 patients (Table S2 and Fig. 2a) [1, 9, 10, 13,14,15,16,17,18,19,20,21,22,23,24,25]. Detailed procedures for the literature review are described in Fig. S3. The median age at the time of diagnosis among the 34 patients was 5 years (range: 0–80 years), including our patient. Among these patients, data on CD19+ B cells were available for 30 patients, with a median CD19+ B-cell count of 6.9 × 109/L (range: 0.6–48.2 × 109/L). The distribution of age and B-cell count among the patients is depicted in Fig. 2b, while the locations of the CARD11 gene mutation are illustrated in Fig. S2b. Out of the 35 patients, 15 presented with mild symptoms or were asymptomatic and did not require immunoglobulin replacement therapy, splenectomy, or immunosuppressive therapy. Among the 21 patients who were 3 years old or older at the time of diagnosis, all achieved long-term survival. However, among the 14 patients who were younger than three years old at diagnosis, six died within 3–9 years of diagnosis. Two of these deaths were due to refractory hemophagocytic lymphohistiocytosis, while the remaining four were caused by opportunistic infections.
Fig. 2
Clinical course of patients with B-cell expansion with nuclear factor kappa B and T-cell anergy (BENTA) identified from a literature review. (a) The swimmer plot illustrates the treatment courses of 34 patients with BENTA. (b) The distribution of age and B-cell count. The gray boxes represent the normal age-related ranges for absolute CD19+ B-cell counts. Out of the 16 patients who were younger than 3 years at the time of diagnosis, six died within 3–9 years following their diagnosis due to refractory hemophagocytic lymphohistiocytosis or opportunistic infections. UPN, unique patient number; SCIG, subcutaneous immunoglobulin; IVIG, intravenous immunoglobulin
Four patients underwent sirolimus treatment. In two cases, the patients were initially suspected of autoimmune lymphoproliferative syndrome because of B-cell proliferation, splenomegaly, anemia, thrombocytopenia, and recurrent respiratory infections [9, 10]. Sirolimus effectively reduced the size of palpable lymph nodes and spleen in these two patients. In another two cases, the patients had concomitant severe multisystem autoimmune diseases [9, 10]. Immunosuppressive combination therapy, including steroids, rituximab, and sirolimus, was employed to treat cytopenia. Despite initial improvement, the course of treatment eventually failed to control the progression of the disease in these two patients.
Two patients underwent allogeneic hematopoietic stem cell transplantation (HSCT). The first patient was a 3-month-old boy who received HSCT from a matched, unrelated donor to address severe immune dysregulation, including cytopenia, granulomatous lymphocytic interstitial lung disease, and nephrotic syndrome [10]. However, he experienced primary graft failure. Consequently, he received a second transplant from the same donor, but unfortunately, he again experienced graft failure and ultimately died due to an adenovirus infection. The second case involved an adult male who received a matched HSCT from his sister to treat chronic lymphocytic leukemia, which developed around the age of 44 [1]. Prior to the diagnosis, he had experienced persistent benign B-cell lymphocytosis since infancy. In adulthood, he presented with a persistent upper respiratory illness and pleural effusion. Following the HSCT, he has remained disease-free for five years.
Comments (0)