
Remember me
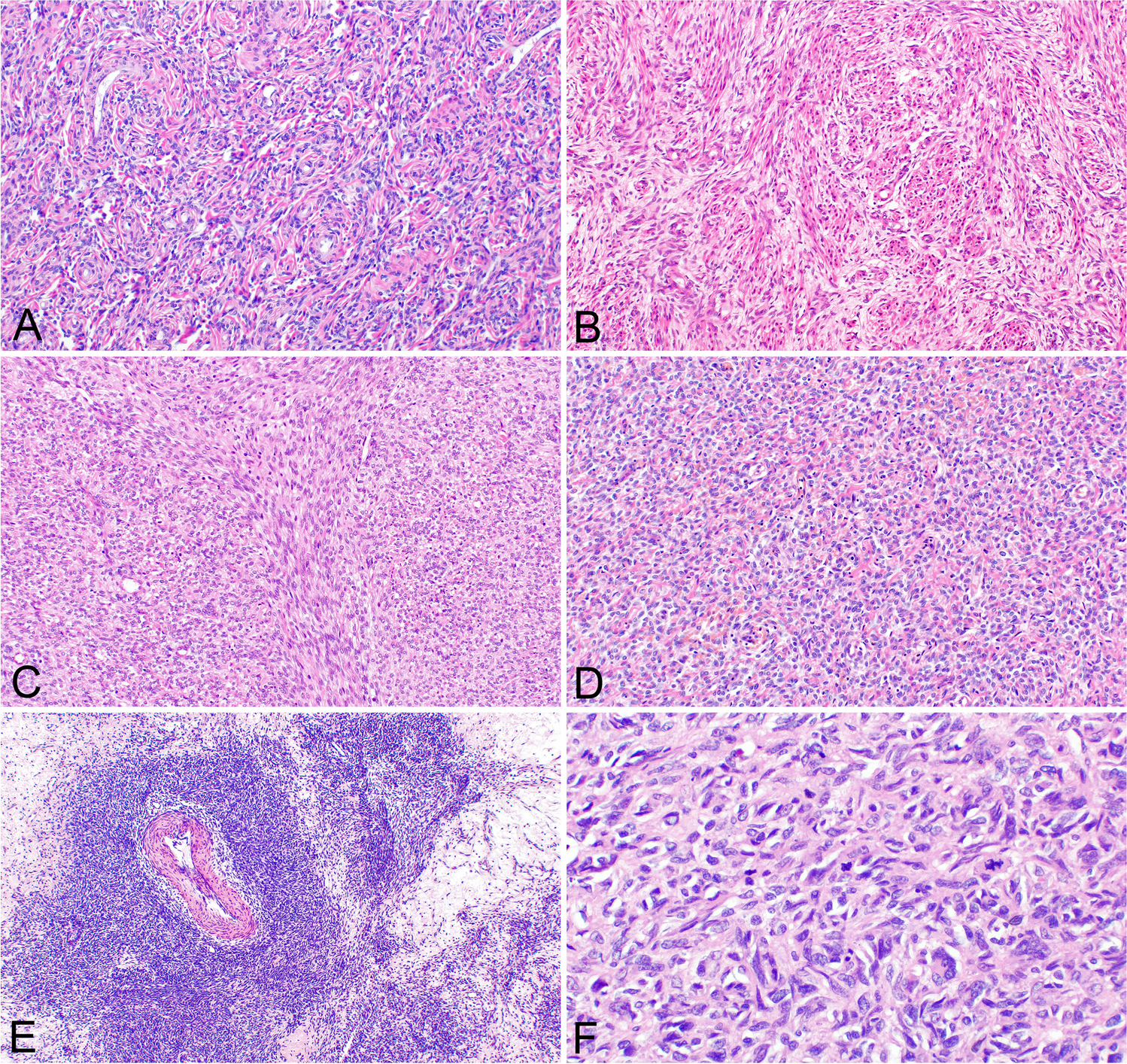

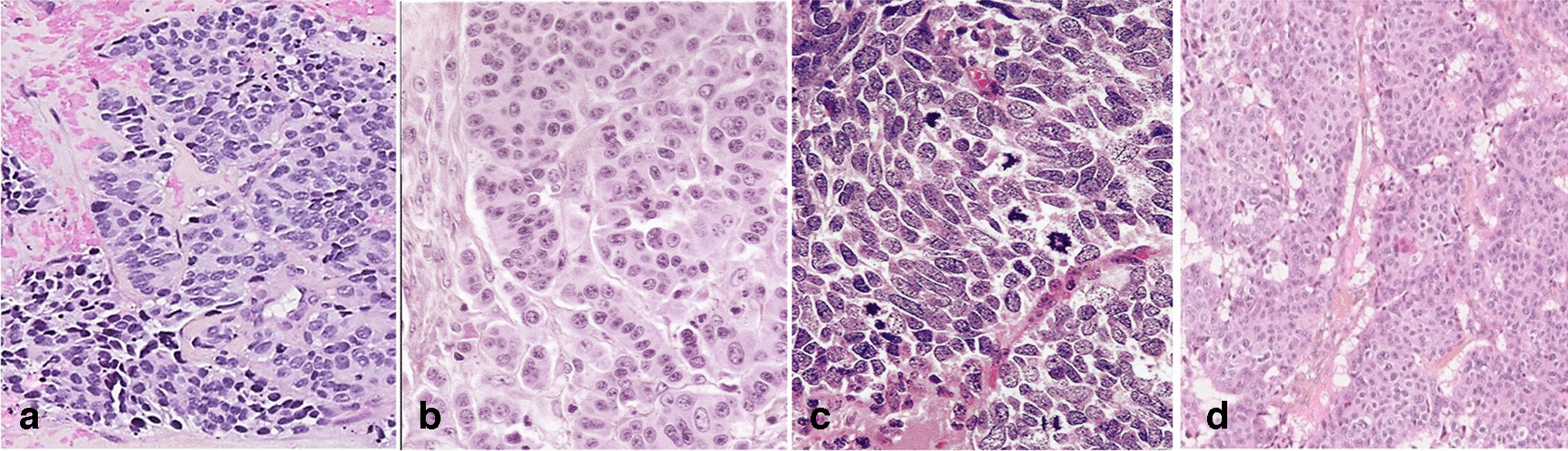
The recurrent-FSGS group showed the highest incidence of the COL variant among the groups, which was even higher when limited to cases with severe proteinuria, especially those within 3 months of transplantation. The pathological characteristics of the COL variant were the cell bridges, where the foot processes and rearrangement of actin filaments could not be confirmed. Glomerular epithelial stem cells (GESCs) near the urinary pole of Bowman’s capsule are thought to have the potential to regenerate podocytes, as identified by the surface markers CD24+ and CD133+ [14, 15]. When podocytes are rapidly lost, GESCs form a cell bridge and promptly replace the lost podocyte (Fig. 4) [14, 16, 17]. The glomerular structure is distorted at the podocyte injury sites, which alters the polarity of GESC division and initiates an abnormal proliferation [14]. These proliferating epithelial cells may be activated parietal epithelial cells (PECs), as they are ectopically positive for PEC markers (Pax-2, Claudin-1, and CK18/8) and CD44 [18]. Epithelial phenotype alterations occur to a much lesser degree in other FSGS variants with podocyte loss [18]. Interestingly, a study using an experimental model reported that activation of PEC prevented leakage of proteins, suggesting that PEC may be protective at the micro level [19]. Furthermore, mild endothelial injury was observed ultrastructurally. Podocytes are the only cells that produce vascular endothelial growth factor (VEGF) in the glomerulus, which is necessary for the survival and function of its endothelial cells. In an vivo study, the loss of podocyte VEGF-A results in thrombotic microangiopathy, whereas its overexpression causes the COL variant of FSGS and endothelial injury [20]. This indicates that the dysregulation of the signaling pathway from podocyte to endothelial cells due to down-regulated VEGF expression might cause the endothelial injury of these lesions (Fig. 4).
Fig. 4
Mechanisms of the development of FSGS lesions in renal allografts. Recurrent FSGS in renal allografts mainly results from podocyte injury caused by humoral factors. The COL variant occurs when activated PECs proliferate and replace podocytes in areas where they have degenerated and detached. The collapsed area may develop segmental sclerosis due to increased matrix. In renal allografts, the hyperfiltration status due to a single kidney may damage the endothelium; CNI-induced arteriolopathy or ABMR may exacerbate this. CNI-FSGS can cause ischemia due to vascular toxicity or direct endothelial cell injury. The endothelial damage caused by ABMR is due to DSA, and the induction of intracapillary inflammation by DSA and plasma protein exudation may lead to CEL and NOS variants. There is crosstalk between endothelial and epithelial cells, which affect each other via cytokines. Furthermore, preexisting interstitial fibrosis may increase podocytes susceptibility to injury and induce FSGS. Thus, both epithelial and endothelial injury contribute to FSGS lesion formation in renal allografts, but the degree of involvement depends on the etiology. FSGS, focal segmental glomerulosclerosis; CNI, calcineurin inhibitor; ABMR, antibody-mediated rejection; COL, collapsing; CEL, cellular; PH, perihilar; NOS, not otherwise specified; GESC, glomerular endothelial stem cell; PEC, parietal epithelial cell; HIF-1α, hypoxia-inducible factor-1α; VEGF, vascular endothelial growth factor; ET-1, endothelin-1; ETRA, endothelin receptor A; ROS, reactive oxygen species; NO, nitric oxide; DSA, donor specific antibody; TCMR, T cell-mediated rejection; IF/TA, interstitial fibrosis and tubular atrophy
The histological variants that observed in native kidneys are similar to that observed in the renal allografts of patients with FSGS. However, the early histologic findings of recurrent FSGS shortly after transplantation are similar to minimal change disease (MCD), i.e., normal light microscopy with moderate/severe foot process effacement on electron microscopy [21]. Subsequently, the detachment of podocytes leads to segmental sclerosis and glomerulosclerosis in patients who are unable to achieve remission [22]. FSGS and MCD are well-known podocytopathies that are also considered to exist along the spectrum of podocyte injury [21]. The present study did not include MCD-like cases. However, similar to previous reports [21, 22], serial biopsy specimens revealed that MCD-like lesions preceded FSGS lesions (Supplemental Fig. 3); this finding may be considered an important clue to the pathogenesis of recurrent FSGS after transplantation.
Mutations in structural podocyte genes cause FSGS with severe nephrotic syndrome at birth or during childhood. Because of their similar clinical presentations, they are often diagnosed as primary FSGS caused by circulating factors, unless genetic testing is performed. No patient in the recurrent-FSGS group had information on genetic abnormalities in their medical records. Genetic cause of FSGS generally do not recur following kidney transplantation as the cause no longer exists. Therefore, if a patient with genetic FSGS develops FSGS post-transplantation, other causes (e.g., CNI toxicity) are implicated.
The CNI-FSGS group demonstrated high percentages of global sclerosis and IF/TA, caused by ischemia, and prominent endothelial cell injury was observed on EM. The glomerular and peritubular capillary endothelial cells are important targets for CNI, and circulating endothelial cells have been reported to be markers of endothelial injury due to CNI toxicity [5]. Conversely, the COL variant was also found in approximately 7% of cases in our study. A possible mechanism for epithelial cell injury in CNI toxicity is podocyte hypoxia due to severe hyaline deposition increasing hypoxia-inducible factor-1α (HIF-1α) with a subsequent increase in VEGF in the podocytes, leading to epithelial hyperplasia [23] (Fig. 4). However, epithelial hyperplasia does not occur in all cases of severe arteriolar hyalinosis, suggesting factors other than ischemia may be involved. Recent studies have demonstrated that accumulated oxidative stress in endothelial cells via the endothelin-1 (ET-1) signaling pathway may be involved in podocyte injury. CNI increases ET-1 in endothelial cells [24], and ET-1 exerts its effects via endothelin receptor A (ETRA) [24] which is localized primarily in renal vasculature smooth muscle cells under normal conditions. However, Daehn et al. reported that ET-1 derived from podocytes activates endothelial cells’ ETRA, causing the accumulation of oxidative stress in endothelial cell mitochondria, which in turn induces podocyte apoptosis (Fig. 4) [25, 26]. An increase in ETRA and oxidative stress in glomerular endothelial cells was also observed in cases of human FSGS [27].
The ABMR-FSGS group showed the highest incidence of the CEL variant among the groups, which had prominent endothelial injury. Endothelial injury in ABMR-related FSGS lesions may be due to the binding of DSA to donor endothelial cell membrane antigen (Fig. 4). DSA can cause indirect injury via complement fixation from classical pathway activation [28], where C4d is the final degradation product of C4 activation and remains stable on the endothelial cell membrane [29]. Conversely, in cases of ABMR with DSA without C4d deposition, DSA can cause direct capillary endothelium injury, or indirect injury via recruitment of inflammatory cells with Fc receptors [28].
In the UE-FSGS group, 72.6% of the cases were of the NOS variant. However, despite the mildest atherosclerosis among the groups, the percentage of IF/TA was the second highest in the CNI-FSGS group, suggesting that nonischemic mechanisms may also be involved. Glomerulonephritis was present in 27.5% of all cases, which was most common in the UE-FSGS group (38.9%). When other types of glomerulonephritis are present, segmental lesions are more easily regarded as their postinflammatory scars. In renal allografts, however, determining the origin of segmental lesions is often difficult because of factors, such as hyperfiltration from a single kidney, drugs, and rejection. Furthermore, the UE-FSGS group included many TCMR or reflux nephropathy cases with prominent IF/TA. Tubular cells secrete growth factors which activate fibroblasts producing an extracellular matrix [30], which causes tubulointerstitial fibrosis and secondary FSGS (Fig. 4). Moreover, a recent report showed that preexisting tubulointerstitial injury makes the glomerulus more susceptible to subsequent podocyte damage (Fig. 4) [31].
We examined semiquantitative urine protein by according to the histological variant in each group (Supplemental Fig. 2). Although it is difficult to draw conclusions, each group exhibited a similar or different trend compared to that reported in native kidneys [13]. We have previously reported that quantitative measurement of several parameters on electron micrographs was helpful in determining the degree of injury in epithelial and endothelial cells in native FSGS cases [8]. While the association between the degree of injury and proteinuria can be evaluated, we were not able to evaluate 24-h proteinuria in the present study. Unlike that performed in native kidney cases, 24-h urine collected to evaluate proteinuria was not performed in many of the renal transplant cases used in the study.
In conclusion, categorizing FSGS lesions in renal allografts by etiology revealed distinct clinicopathologic features, whereas it is difficult to identify the etiology from histological variants. This is because even in the same FSGS variants, there were differences in the degree of epithelial and endothelial injury, which may reflect different segmental lesion mechanisms. For example, the COL variant may occur by podocyte injury and also by endothelial injury or disruption of epithelium and endothelium crosstalk, which may relate to the molecular mechanisms involved, treatment and prognosis. Precise observation of FSGS lesions and understanding the pathogenesis of the segmental lesions may help in the diagnosis and clinical management of FSGS during renal transplantation.
Comments (0)