
Remember me
A 77-year-old male presented with intermittent abdominal pain. CT scan revealed a 4.7 cm retroperitoneal mass. A subsequent fine-needle aspiration (FNA)with a core biopsy revealed basophilic clumps of acellular homogenous material on Diff Quik stains performed during rapid on site evaluation [Figure 1a]. Pap stain showed greenish round deposits [Figure 1b], whereas cell block showed dense hyalinization and amorphous eosinophilic material amidst benign liver parenchyma [Figure 1c and d].
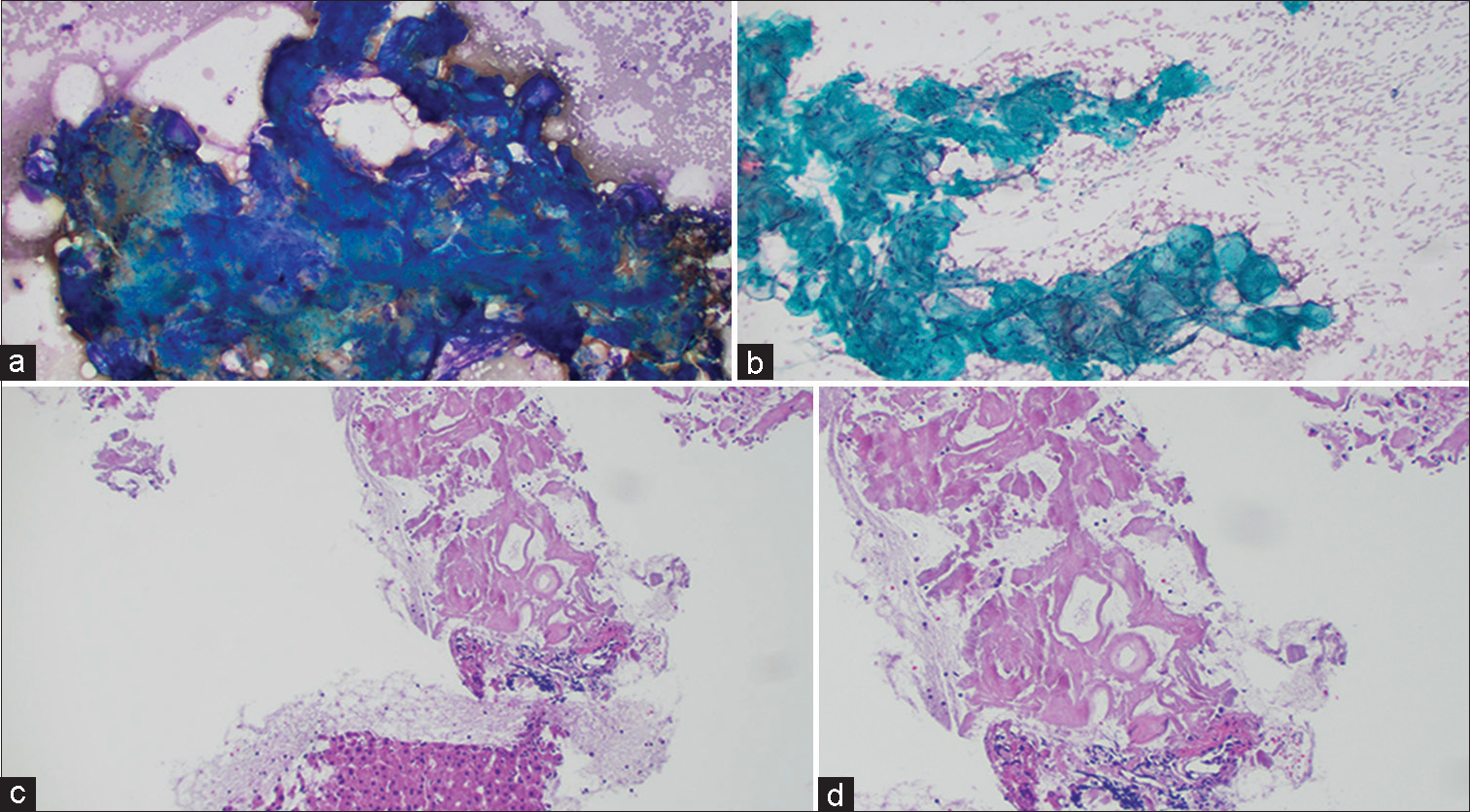
Figure 1:: (a) Retroperitoneal fine-needle aspiration (FNA): Diff Quik Stain showing acellular, blue, amorphous material. (b) Retroperitoneal FNA: Pap stain showing acellular, greenish, rounded amorphous material. (c) Retroperitoneal FNA: Cell block with H and E stain showing hyaline deposits of amorphous eosinophilic material, amidst benign liver parenchyma (Low power view). (d) Retroperitoneal FNA: Cell block with H and E stain showing hyaline deposits of amorphous eosinophilic material (High power view).
Export to PPT
What is the diagnosis?
Soft-tissue tumor
Solitary fibrous tumor
Collagenous spherulosis
Amyloid
Answer:
d) Amyloid
The most common diagnosis of retroperitoneal masses is a soft-tissue tumor such as a liposarcoma. Other common entities that can lead to retroperitoneal mass formation are lymphomas. Collagenous nodules as seen in our patient on H and E section can be also seen in some cases of retroperitoneal fibrosis. However, in our patient, the collagenous areas were striking and amorphous and immediately raised the possibility of amyloid deposits. The presence of inflammatory cells and plasma cells also favored the diagnosis of amyloid.
The diagnosis was confirmed by positive Congo staining [Figure 2] with apple green birefringence [Figure 3]. Serum protein electrophoresis and immunofixation were negative, but his serum lambda-free light chains were elevated at 109.3 mg/L with a low of kappa/lambda of 0.21 prompting referral to hematology. Liquid chromatography-tandem mass spectrometry was performed on peptides extracted from micro dissected areas of the paraffin-embedded specimen and detected a peptide profile consistent with amyloid light chain (AL) (lambda)-type amyloid deposition, supporting the diagnosis of AL amyloidosis. The patient’s abdominal pain had subsided, and bone marrow biopsy was deferred for later. There was not enough evidence to determine if the amyloidosis was systemic or localized.
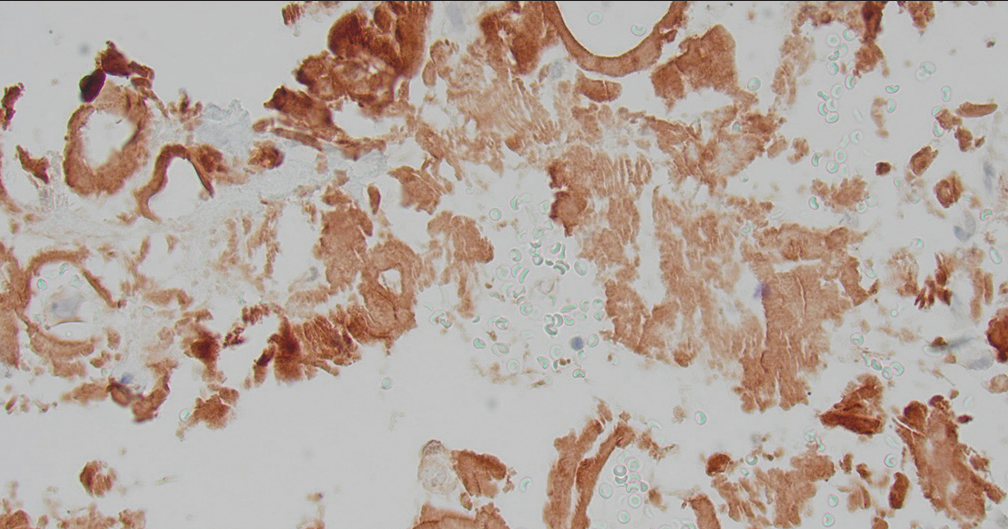
Figure 2:: Retroperitoneal fine-needle aspiration: Congo red stain showing brownish red acellular, amorphous deposits.
Export to PPT
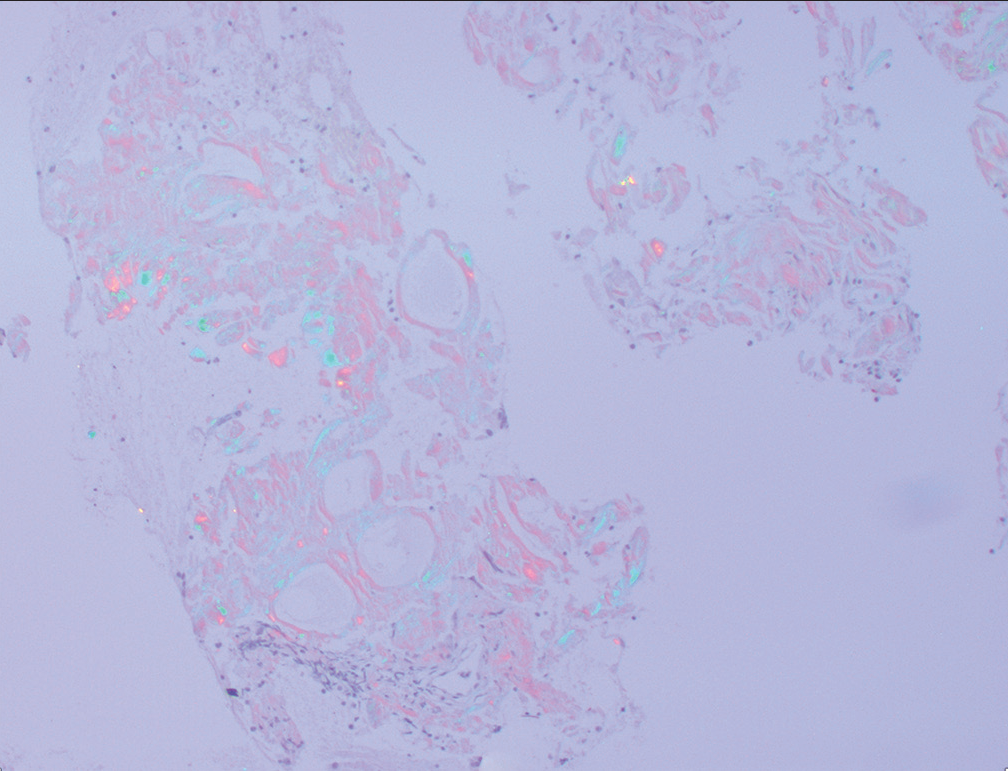
Figure 3:: Retroperitoneal fine-needle aspiration: Congo red stain showing apple green birefringence under polarized light.
Export to PPT
Amyloidosis is a disease that occurs from the deposition of mis-folded proteins in organs and tissues. Amyloid refers to a group of abnormal proteins that share a ß-pleated sheet structure.[1] This structure makes the proteins insoluble and unsusceptible to enzymatic digestion as well as responsible for its physical attributes such as its amorphous homogenous eosinophilic material and “apple-green” birefringence of Congo red stains under polarized light.[1]
There are different types of amyloid deposits, but AL (A = amyloid, L = light chain) is the most common and is usually seen to affect older men. In AL amyloidosis, plasma cells produce unstable quantities of monoclonal free light chains.[2] AL amyloidosis can be of the kappa or lambda type. These free light chains join to form amyloid fibrils, which the body cannot eliminate easily.[2] Amyloid fibrils start to build up in tissues and organs, inhibiting their function over time. Amyloids can precipitate in the same location, in which they are created resulting in localized amyloidosis. Amyloids can also spread throughout the bloodstream to other organs resulting in systemic amyloidosis.[3]
Which new specific and sensitive test can confirm the presence of amyloid and also specify the type (AA, AL [Kappa or Lambda], and transthyretin/hereditary type)?
Congo red staining
Immunohistochemistry
Liquid chromatography-tandem mass spectrometry
Electron microscopy
Answer(s):
c) Liquid chromatography-tandem mass spectrometry
AL amyloidosis is typically diagnosed by staining the sample with Congo red and observing for apple-green birefringence. Immunohistochemistry is not useful as it often results in non-specific staining and the sensitivity of the technique is low. Using this process, diagnosis of the specific kind of amyloidosis and determining if it is systemic or localized is difficult. However, technological advancements in mass spectrometry have made it more efficient for diagnosing amyloidosis. Mass spectrometry can analyze specific proteins allowing for a diagnosis of not only amyloidosis but also the type of amyloidosis, giving more options of treatment to the patient. Findings from mass spectrometry can also provide a general diagnosis independent of the application of Congo red staining.[4] In addition, mass spectrometry can be easily performed on formalin fixed paraffin-embedded tissue.[4]
ADDITIONAL QUESTIONSWhat are the symptoms of AL amyloidosis?
Shortness of breath
Chronic kidney disease
Constipation
Arrhythmia
All of the above
Answer(s):
d. All of the above
AL amyloidosis is generally found in older men. General symptoms of AL amyloidosis include abnormal bleeding, dizziness, fatigue, shortness of breath, and muscle weakness.[5] AL amyloidosis also hinders the function of specific organs. A build-up of amyloid proteins in the kidneys can cause chronic kidney disease (renal AL amyloidosis).[5] Accumulation of amyloid proteins in the heart (AL cardiac amyloidosis) can cause heart palpitations, arrhythmia, shortness of breath, fatigue, as well as thickening of heart tissues increasing the chance of heart failure.[5] In the gastrointestinal tract, escalation in amyloid proteins can cause weight loss, abdominal pain, constipation, diarrhea, gastric reflux, and vomiting.[5] Amyloid proteins can also develop in the central nervous system causing changes in blood pressure, erectile dysfunction, pain in the hands, feet, and lower legs, as well as confusion.[5] Unlike some forms of amyloidosis, AL amyloidosis cannot be transmitted genetically.[3]
What treatments are implicated on a patient with AL amyloidosis?
Chemotherapy
No treatment
Bone marrow transplant
Removal of the mass
All of the above
Answer(s):
e). All of the above
There is no known cure for systemic AL amyloidosis. Chemotherapy, like that used in cancer patients, is typically prescribed to slow down the build-up of amyloid proteins by killing cells that produce them.[6] Another method is bone marrow transplant, also known as a stem cell transplant. Patients with localized amyloidosis do not receive treatment, instead, the mass is removed if it affects organ function; otherwise, the mass can be left in the body.
What is the latest method of confirming a clinical diagnosis of amyloidosis?
Blood test
Fat pad biopsy with Congo red staining and electron microscopy
Anterior fat pad FNA biopsies with mass spectrometry/ proteomics
Rectal biopsies
Answer:
c). Anterior fat pad FNA biopsies with mass spectrometry/ proteomics
Earlier, abdominal fat biopsies for confirming amyloidosis relied on electron microscopy and Congo red staining.[7] Now, better tests for diagnosis are available, and FNA samples can be aspirated successfully[8,9] and processed using laser dissection, mass spectrometry, and proteomics.[10] Rectal mucosal biopsies have been replaced by anterior abdominal wall fat pad FNA technique as shown in videos.[8,9]
SUMMARYAL amyloidosis is a rare condition typically found in older male patients. Initial diagnosis of AL amyloidosis usually involves Congo red staining, looking for apple-green birefringence. Mass spectrometry recognizes all amyloid types (AL vs. AA vs. transthyretin/hereditary) and can easily be performed on formalin fixed, paraffin embedded tissue from core biopsies, and cell blocks. It confirms the diagnosis, identifies specific mutations, and helps guide appropriate treatment.
Comments (0)