
Remember me
Tuberculosis (TB), one of the oldest diseases known to affect humans, is caused by the Mycobacterium tuberculosis (MTB) complex.[1,2] Every year, around 10 million people are infected with TB worldwide, and about 1/4 of the population develops latent MTB infection.[3,4] Despite vaccines and chemotherapy, patients still develop latent TB infections. The interaction between macrophages and MTB plays an important role in TB progression. MTB can also manipulate macrophage defense mechanisms to maintain its own survival during the course of TB.[5] Activated macrophages are related to the prognosis of pulmonary TB.[6] Therefore, the potential molecular mechanisms affecting MTB-infected macrophage functions must be urgently explored to provide a therapy target for TB.
The switch/sucrose non-fermenting (SWI/SNF) family is an essential chromatin regulator,[7] and its subunit mutations cause a variety of different developmental disorders in humans.[8] SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 (SMARCA5) is a core adenosine triphosphatase belonging to the SWI/SNF family[9] and has an important role in human disease progression. SMARCA5 reprograms aldoketo reductase family 1 member B1-mediated fructose metabolism to regulate leukemia progression.[10] Jin et al.[11] found that SMARCA5 is upregulated in breast cancer, and its downregulation could inhibit cancer cell proliferation and invasion. In a TB-related study, SMARCA5 expression was increased in Bacillus Calmette–Guérin (BCG)-induced Human Promyelocytic Leukemia Cell Line (THP-1) cells, and its downregulation promoted BCG-induced THP-1 cell viability and inhibited apoptosis, suggesting that SMARCA5 might suppress mycobacterial-infected macrophage growth to promote TB progression.[12] Whether SMARCA5 regulates MTB-induced macrophage polarization remains unclear.
TB susceptibility is associated with epigenetic changes in the genes involved in immune and inflammatory responses.[13] m6A methylation, one of the most common RNA modifications, can affect many RNA processes, including splicing, translation, output, and decay.[14] m6A methylation is catalyzed by methyltransferase 3 (METTL3), whose residues can be recognized by m6A reader insulin-like growth factor 2 binding protein 1/2/3 (IGF2BP1/2/3).[15] METTL3 promotes cell death and inflammation in MTB-infected macrophages, thus aggravating TB progression.[16] In this study, we discovered the presence of methylation modification sites in SMARCA5 using the SRAMP website prediction and found evidence on the ENCORI database that METTL3 might interact with SMARCA5. However, whether METTL3 affects the m6A methylation of SMARCA5 remains unexplored.
Here, we speculated that the METTL3-mediated m6A modification of SMARCA5 regulates MTB-induced macrophage polarization and inflammation. Our study may provide new ideas for developing TB treatment strategies.
MATERIAL AND METHODS Cell culture and transfectionTHP-1 cells (TIB-202; ATCC, Manassas, VA, USA) were grown in Roswell Park Memorial Institute-1640 medium (PM150110, Procell, Wuhan, China) containing 10% Fetal Bovine Serum,164210-50, Procell) and 1% penicillin/streptomycin (PB180120, Procell) and treated with phorbol myristate acetate (100 ng/mL; P1585, Sigma–Aldrich, St Louis, MO, USA) to induce a macrophage-like state. THP-1 cells were identified by Short Tandem Repeat, and polymerase chain reaction (PCR) Mycoplasma Test Kit (WLA111a, Wanleibio, Shenyang, China) was used to confirm that the Mycoplasma was negative in THP-1 cells. To mimic TB cell models, THP-1 macrophages were treated with MTB at a multiplicity of infection (MOI) of 0, 1, 5, and 10 for 24 h. In accordance with the instructions of Lipofectamine 3000 (L3000075, Invitrogen, Waltham, MA, USA), the cells were transfected with small interfering RNAs (siRNAs) (RiboBio, Guangzhou, China) for SMARCA5/METTL3/IGF2BP1/IGF2BP2/IGF2BP3/YTH domain-containing family (YTHDF)1/YTHDF2/YTHDF3 (siSMARCA5: F 5ʹ-AUUUUUUCUUCAUAGGUAGGA-3ʹ, R 5ʹ-CUACCUAUGAAGAAAAAAUGC-3ʹ; siMETTL3: F 5ʹ-UCUAACUCAGGAUCUGUAGCU-3ʹ, R 5ʹ-CUACAGAUCCUGAGUUAGAGA-3ʹ; siIGF2BP1: F 5ʹ-AUGUAAAGCUUGUUCAUGGUG-3ʹ, R 5ʹ-CCAUGAACAAGCUUUACAUCG-3ʹ; si-IGF2BP2: F 5ʹ-ACACAAUACCGGUCAUUAGCU-3ʹ, R 5ʹ-CUAAUGACCGGUAUUGUGUAC-3ʹ; si-IGF2BP3: F 5ʹ-AAAAACUACUUUUUGUCUCUU-3ʹ, R 5ʹ-GAGACAAAAAGUAGUUUUUUC-3ʹ; si-YTHDF1: F 5ʹ-UUAUCUUGUCCUUUUGUUCUC-3ʹ, R 5ʹ-GAACAAAAGGACAAGAUAAUA-3ʹ; si-YTHDF2: F 5ʹ-UCCUUUUGAUGUACAGAUCCA-3ʹ, R 5ʹ-GAUCUGUACAUCAAAAGGAUG-3ʹ; si-YTHDF3: F 5ʹ-UAUUUCCUUGCCCUUUAGGUC-3ʹ, R 5ʹ-CCUAAAGGGCAAGGAAAUAAA-3’), and their negative controls. The polyclonal DNA (pcDNA) SMARCA5 or IGF2BP1 overexpression vector was structured by inserting the PCR products of SMARCA5 or IGF2BP1 into pcDNA3.1 (V79020, Invitrogen). After transfection for 24 h, the THP-1 macrophages were infected with 5 MOI MTB for 24 h.
Cell counting kit 8 (CCK8) assayCCK8 Kit (C0041, Beyotime, Shanghai, China) was used to test cell viability. The macrophages in 96-well plates were incubated with 10 μL of CCK8 reagent for 4 h. The absorbance at 450 nm was analyzed using a microplate reader (SpectraMax i3x, Molecular Devices, LLC, Sunnyvale, CA, USA) to assess cell viability.
Flow cytometryThe macrophages were resuspended with binding buffer, mixed with annexin V- fluorescein isothiocyanate (FITC) and propidium iodide staining solution (40302ES, Yeasen, Shanghai, China), and further incubated for 15 min. For cell surface antibody detection, the cells were labeled with phycoerythrin/Cy7® Anti-CD86+ (ab233571, Abcam, Cambridge, MA, USA) for 30 min. After washing, cell apoptosis rate and CD86+-positive cell ratio were assessed by flow cytometry (FACSCanto II, BD Biosciences, San Jose, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)Macrophage lysates were sonicated and centrifuged, and the supernatant was extracted. Commercial kits were used to assess the levels of inflammatory factors. The cell supernatant was incubated with a human interleukin-1 beta (IL-1β) ELISA kit (ab214025, Abcam) and tumor necrosis factor-alpha (TNF-α) ELISA kit (ab181421, Abcam) to evaluate IL-1β and TNF-α levels, respectively, in accordance with the kits’ instructions.
Western blot (WB)Proteins were extracted from macrophages using Radio-Immunoprecipitation Assay buffer (P0013B, Beyotime). After quantification, the protein samples were separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred onto Polyvinylidene Fluoride membranes (FFP32, Beyotime). Afterward, the membranes were blocked and incubated with the following antibodies: Anti-SMARCA5 (1/5000, ab183730, Abcam), anti-METTL3 (1/1000, ab195352, Abcam), anti-IGF2BP1 (1/1000, ab184305, Abcam), anti-glyceraldehyde-3-phosphate dehydrogenase (1/2500, ab9485, Abcam), and goat anti-rabbit immunoglobulin G (IgG) (1/50000, ab205718, Abcam). Protein signals were exposed with BeyoECL Star (P0018AS, Beyotime) and analyzed with Image J software (v1.8.0.345, National Institutes of Health, Bethesda, MD, USA).
m6A level detectionAn m6A RNA methylation assay kit (ab185912, Abcam) was used to test the m6A levels of macrophages. Total RNAs were extracted from the macrophages treated with or without MTB using the TRIzol reagent (15596018, Invitrogen). The samples were incubated with a binding solution for RNA binding. After washing, the cells were incubated with antibodies, detection antibodies, and an enhancer solution. After the reaction was stopped, absorbance values were detected at 450 nm to assess the m6A level.
Quantitative real-time PCR (qRT-PCR)Total RNAs were extracted from macrophages using the TRIzol reagent (15596026CN, Invitrogen), followed by use for complementary DNA (cDNA) synthesis using the PrimeScript Reverse Transcription (RT) reagent kit (RR716, Takara, Tokyo, Japan). SYBR Green (RR820A, Takara) was applied for PCR amplification with cDNA and the following primers: SMARCA5, F 5ʹ-CTTCAGGCTAT GGACCGAGC-3ʹ, R 5ʹ-AGCCTCCCTTGTTGAATGACT-3ʹ; METTL3, F 5ʹ-CAGAGGCAGCATTGTCTCCA-3ʹ, R 5ʹ-ATGGACACAGCATCAGTGGG-3ʹ; IGF2BP1, F 5ʹ-GG ACCGAAGGGAAGAAGCTG-3ʹ, R 5ʹ-GTTT CGATGGCCTTCATCGC-3ʹ; and GAPDH, F 5ʹ-AGAAGG CTGGGGCTCATTTG-3ʹ, R 5ʹ-AGGGGCCATCCACAG TCTTC-3ʹ. The fold change of SMARCA5 was calculated by the 2−ΔΔCt method.
Methylated RNA immunoprecipitation (MeRIP) assayThe SRAMP website (www.cuilab.cn/sramp/) was used to predict the methylation modification sites in SMARCA5, and the ENCORI database was utilized to analyze the interaction between METTL3 and SMARCA5. A messenger RNA (mRNA) purification kit (61006, Invitrogen) was used to purify total RNA from the macrophages, and fragmented RNAs were obtained using fragmentation reagents (AM8740, Invitrogen). In accordance with the Magna MeRIP m6A Kit (17-10499, Millipore, Billerica, MA, USA) instructions, the fragmented RNAs were incubated with anti-m6A or anti-IgG pre-coated with A/G magnetic beads overnight. Afterward, qRT-PCR was performed to calculate the m6A level of SMARCA5 in the cell samples.
RNA immunoprecipitation (RIP) assayAn RIP kit (17-700, Millipore) was used to perform the RIP assay. The transfected macrophages were lysed with RIP buffer, and cell lysates were incubated with anti-IGF2BP1 (ab184305, Abcam) or anti-IgG (ab172730, Abcam) pre-coated with magnetic beads overnight. The complexes were then washed, and RNA was extracted for the detection of SMARCA5 enrichment by qRT-PCR.
mRNA stability assayTransfected macrophages were treated with 10 μg/mL actinomycin D (HY-17559, MCE, Monmouth Junction, NJ, USA) at indicated time points (0, 3, 6, and 9 h). qRT-PCR was performed to measure SMARCA5 mRNA expression.
Statistical analysisBefore statistical analysis, normality tests were performed using the Shapiro-Wilk normality test to confirm that all data were normally distributed. Data were shown as mean ± standard deviation using GraphPad Prism 8.0 software (GraphPad, La Jolla, CA, USA). Statistical significance was analyzed by Student’s t-test (for two groups) or analysis of variance and subsequent Tukey post hoc test (for multiple groups). P < 0.05 indicated statistical significance.
RESULTS MTB-induced macrophage polarization and inflammationBy constructing cell models in vitro, we found that MTB reduced macrophage viability and promoted apoptosis in a concentration-dependent manner (P < 0.05) [Figure 1a and b]. The IL-1β and TNF-α levels in the macrophages increased with the MTB concentration (P < 0.05) [Figure 1c]. The cell ratio of M1 polarization marker CD86+ dose-dependently increased in the MTB-induced macrophages (P < 0.05) [Figure 1d]. These results showed that the MTB treatment contributed to macrophage M1 polarization and inflammation.
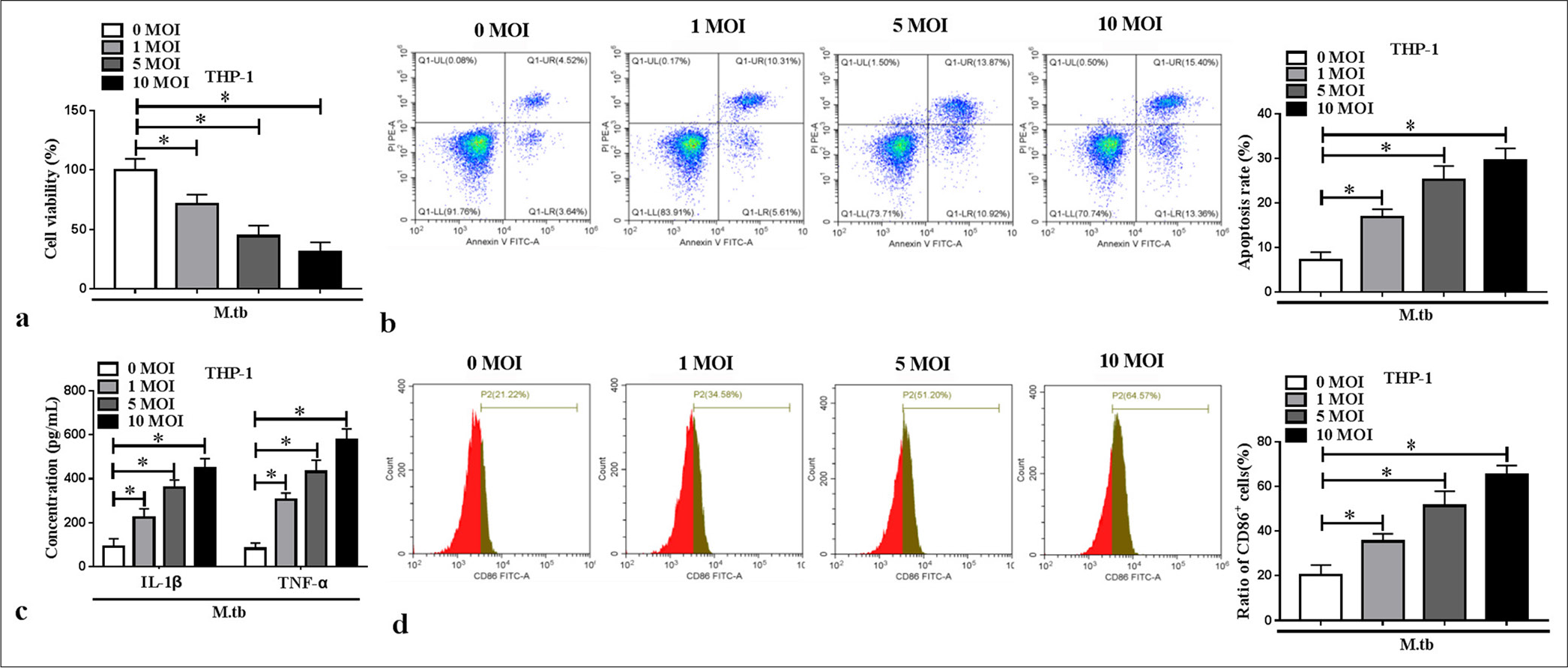
Export to PPT
Silencing of SMARCA5 inhibited MTB-induced macrophage M1 polarization and inflammationConsidering previous findings,[12] we investigated the role of SMARCA5 in MTB-induced macrophage polarization and inflammation. SMARCA5 protein expression concentration dependently increased in the MTB-induced macrophages (P < 0.05) [Figure 2a]. We then transfected si-SMARCA5 into the macrophages to decrease SMARCA5 mRNA and protein expression (P < 0.05) [Figure 2b and c]. Functional experiment results revealed that si-SMARCA5 promoted viability and repressed apoptosis in the MTB-induced macrophages (P < 0.05) [Figure 2d and e]. Meanwhile, SMARCA5 knockdown suppressed IL-1β and TNF-α levels and CD86+ cell ratio in the -induced macrophages (P < 0.05) [Figure 2f and g]. The above findings demonstrated that SMARCA5 might enhance MTB-induced macrophage M1 polarization and inflammation.
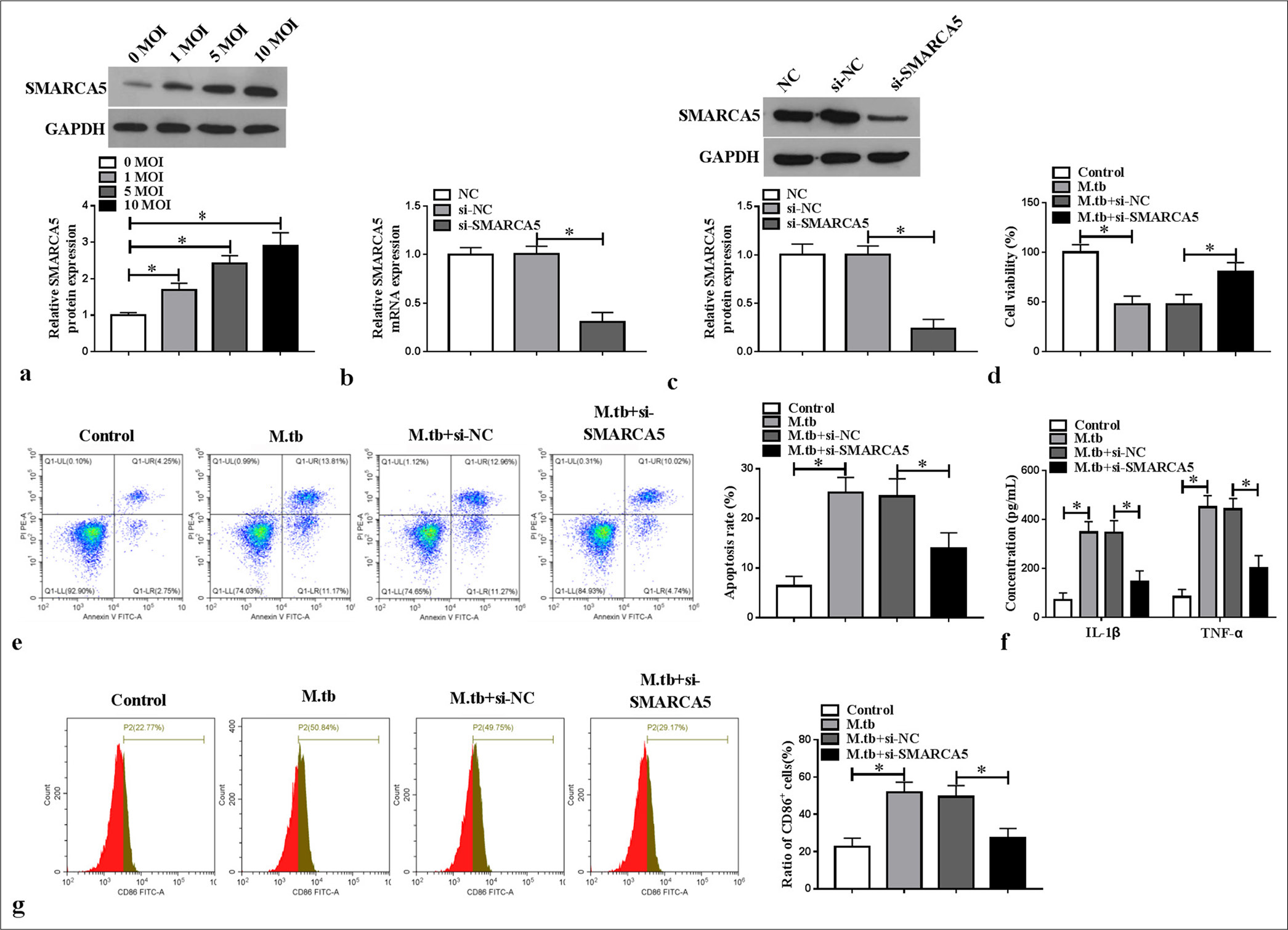
Export to PPT
METTL3 mediated the m6A methylation of SMARCA5Through detection, we found that the m6A level was elevated in the MTB-induced macrophages (P < 0.05) [Figure 3a]. SRAMP website predicted the existence of methylation modification sites in SMARCA5 [Figure 3b], and further MeRIP assay confirmed that SMARCA5 enrichment was increased by anti-m6A in the MTB-induced macrophages (P < 0.05) [Figure 3c]. According to the ENCORI database, METTL3 could interact with SMARCA5 [Figure 3d]. METTL3 protein expression was elevated in the MTB-induced macrophages (P < 0.05) [Figure 3e]. For further analysis, si-METTL3 was constructed to reduce METTL3 mRNA and protein expression in the macrophages (P < 0.05) [Figure 3f and g]. We found that SMARCA5 mRNA expression was decreased by si-METTL3 in the macrophages (P < 0.05) [Figure 3h]. Furthermore, METTL3 knockdown inhibited the m6A level of SMARCA5 in the MTB-induced macrophages (P < 0.05) [Figure 3i], and METTL3 silencing reduced the mRNA stability of SMARCA5 (P < 0.05) [Figure 3j]. These findings indicated that METTL3 enhanced SMARCA5 expression by promoting the latter’s m6A methylation.
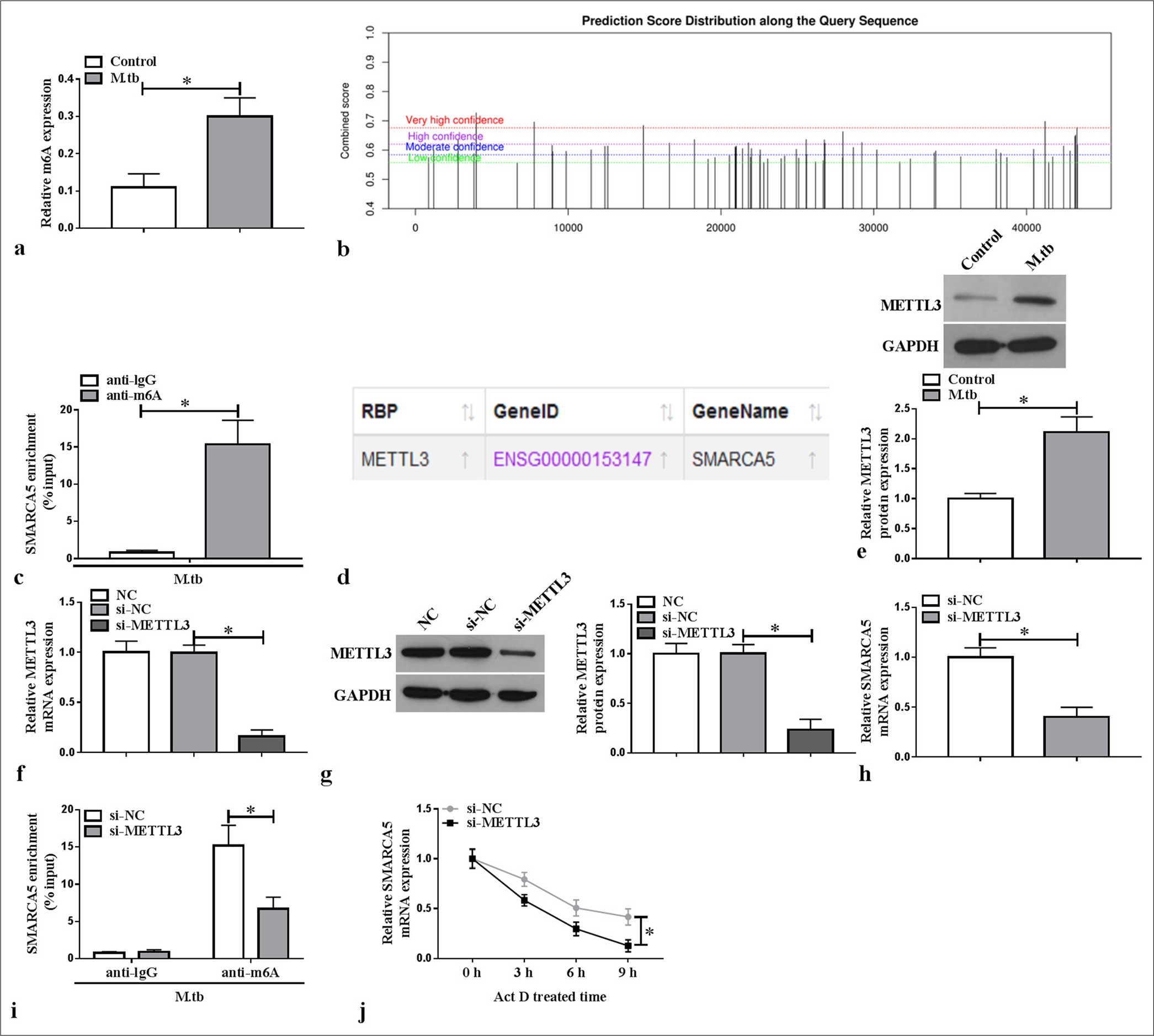
Export to PPT
IGF2BP1 recognized the METTL3-mediated m6A methylation of SMARCA5We constructed the siRNAs for m6A reader families and confirmed the knockdown efficiency of si-IGF2BP1/si-IGF2BP2/si-IGF2BP3/si-YTHDF1/si-YTHDF2/si-YTHDF3 using WB analysis (P < 0.05) [Supplementary Figure 1a-f]. We also tested the effect of m6A reader families on SMARCA5 expression by qRT-PCR and found that only si-IGF2BP1 reduced SMARCA5 mRNA expression in the MTB-induced macrophages (P < 0.05) [Figure 4a]. SMARCA5 enrichment was increased by anti-IGF2BP1 in the MTB-induced macrophages, and METTL3 knockdown could abolish this effect (P < 0.05) [Figure 4b]. This finding confirmed that METTL3 affected the interaction between SMARCA5 and IGF2BP2. Furthermore, IGF2BP1 mRNA and protein expression levels were increased in the macrophages transfected with the IGF2BP1 overexpression vector (P < 0.05) [Figure 4c and d]. IGF2BP1 overexpression reverted the si-METTL3-mediated inhibition of SMARCA5 mRNA expression in the macrophages (P < 0.05) [Figure 4e]. These results illustrated that the METTL3-mediated m6A methylation of SMARCA5 could be directly recognized by IGF2BP1.
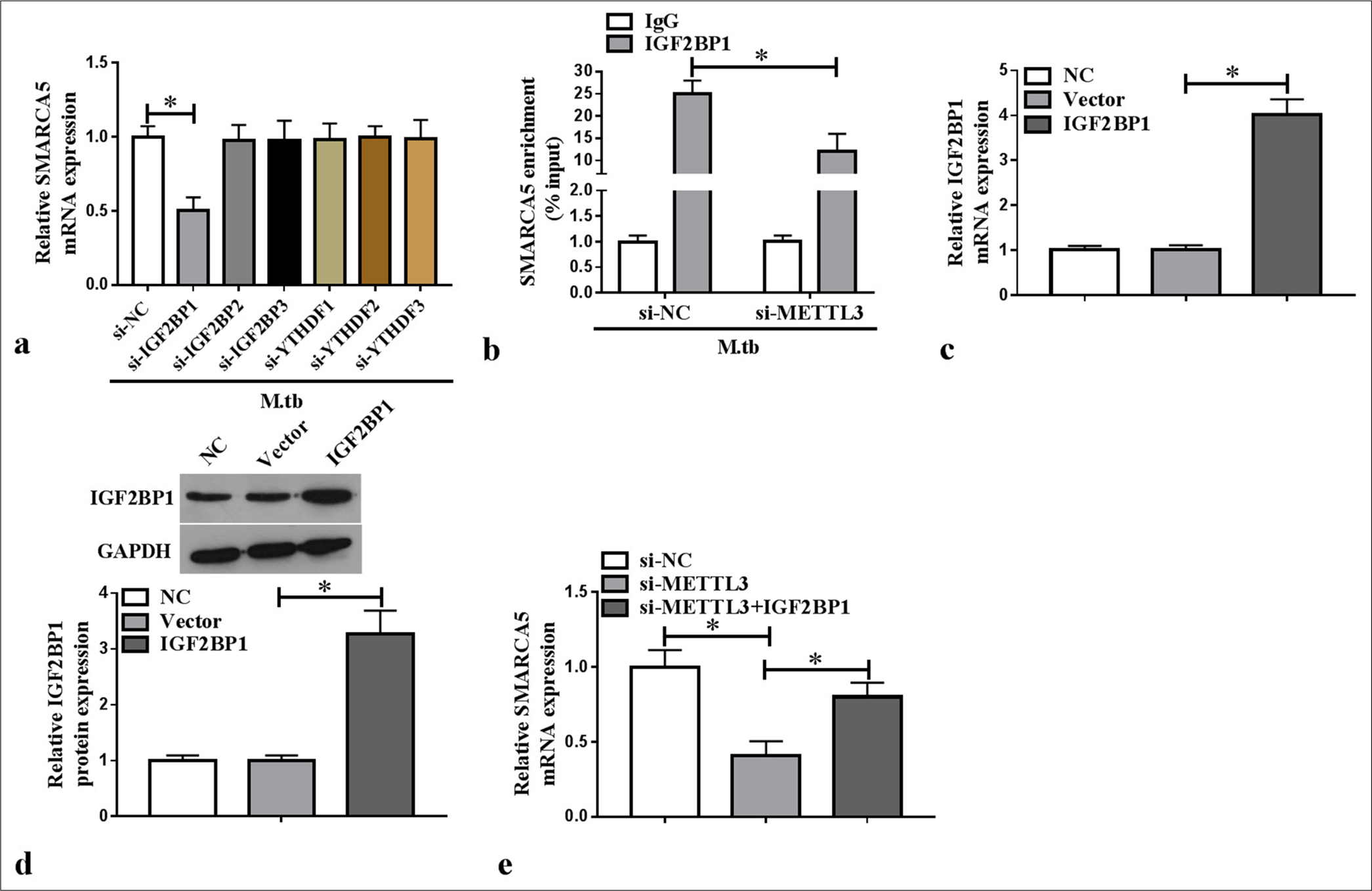
Export to PPT
Overexpression of SMARCA5 reversed the si-METTL3-mediated repression of M1 polarization and inflammationThe mRNA and protein expression levels of SMARCA5 were upregulated in the macrophages transfected with SMARCA5 overexpression vector (P < 0.05) [Figure 5a and b]. Analysis showed that METTL3 knockdown inhibited viability and increased the apoptosis in the MTB-induced macrophages, and these effects were abolished by SMARCA5 overexpression (P < 0.05) [Figure 5c and d]. SMARCA5 overexpression reversed the repression effect of si-METTL3 on IL-1β, TNF-α, and CD86+ cell ratio in the MTB-induced macrophages (P < 0.05) [Figure 5e and f]. Overall, METTL3 enhanced the MTB-induced macrophage M1 polarization and inflammation by upregulating SMARCA5.
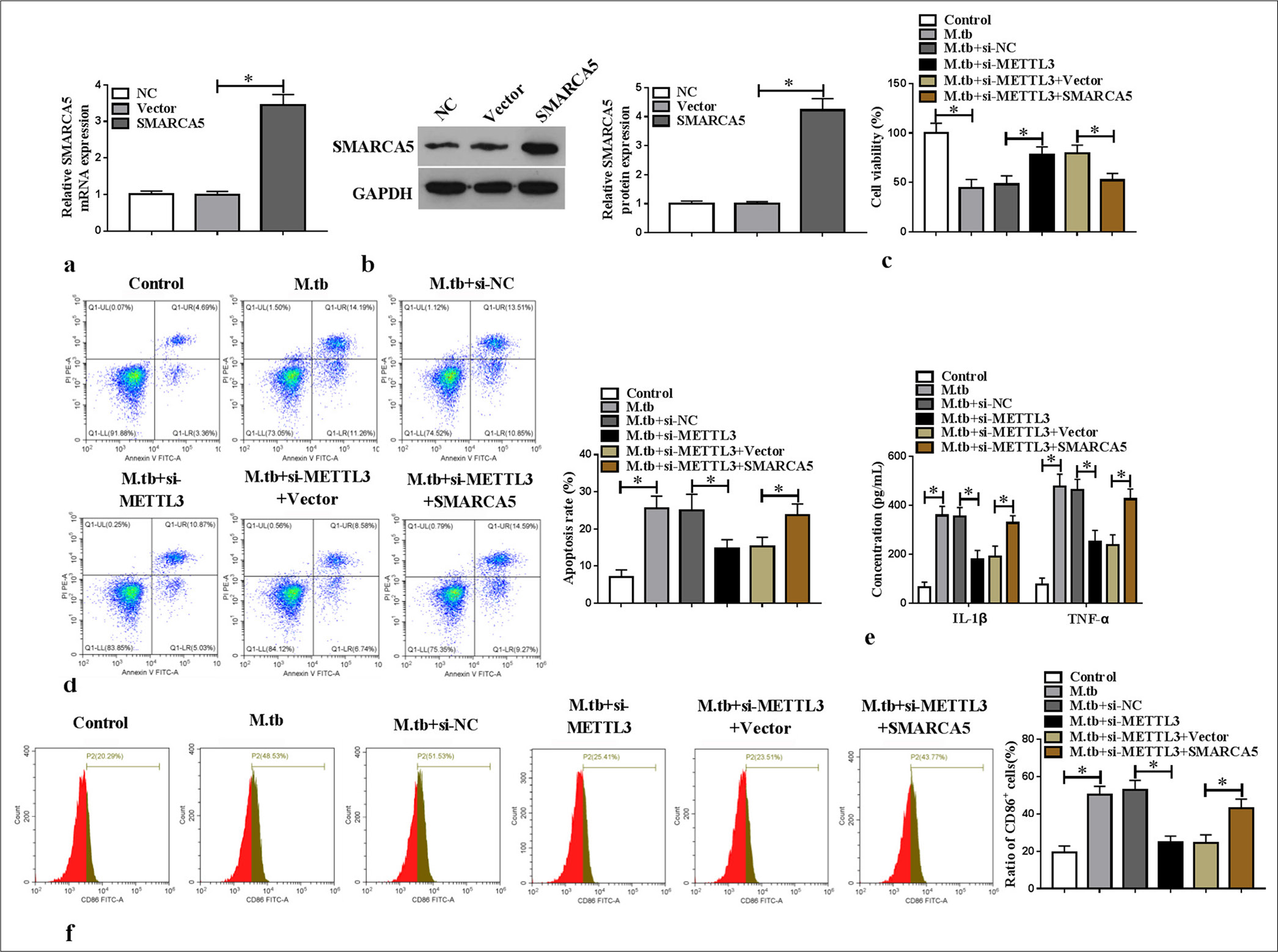
Export to PPT
DISCUSSIONTB remains a major unsolved public health threat worldwide, killing 1 million people each year. MTB is a pathogen of TB that primarily infects, colonizes, survives, and grows inside macrophages; these two interact and affect each other inside the body.[17] MTB disrupts the immune function of macrophages and establishes infection by regulating macrophage metabolism.[18] The molecular and cellular functions of MTB response can be altered by modulating macrophages.[19] This study focused on exploring the potential molecular mechanisms that influence macrophage functions in MTB infection. Our findings showed that the METTL3/SMARCA5 axis promoted the M1 polarization and inflammation of MTB-infected macrophages, providing a new target for TB treatment.
Adenosine triphosphate-dependent chromatin remodeling protein SMARCA5 is necessary to control gene expression and DNA replication and repair.[9,20] SMARCA5 plays an important role in a variety of diseases, such as aggravating ulcerative colitis.[21] MiR-146a represses the proliferation and invasion of glioblastoma patient-derived primary cells by downregulating SMARCA5.[22] Meanwhile, SMARCA5 is upregulated in high-grade gliomas, and its knockdown inhibits cell proliferation and metastasis.[23] Although previous studies have revealed that SMARCA5 may mediate TB progression by regulating the function of BCG-infected macrophages,[12] the roles and molecular mechanisms of SMARCA5 in TB still require further exploration. In this study, we found that MTB infection could repress the viability and enhance the apoptosis and M1 polarization and inflammation in macrophages, indicating the successful construction of the TB cell models. We observed an increase in SMARCA5 expression, suggesting that SMARCA5 might participate in TB progression. Loss-of-functional experiments revealed that SMARCA5 knockdown eliminated MTB-induced macrophage M1 polarization and inflammation, confirming that SMARCA5 might be an effective target for TB.
m6A is the most abundant mRNA modifier in eukaryotes.[24] Increasing evidence indicates that METTL3, dependent or independent of its m6A methyltransferase activity, plays a key role in multiple diseases.[15] The m6A level is elevated in gastric cancer, and METTL3-mediated HDGF enhances gastric cancer proliferation and liver metastasis.[25] m6A readers, including the YTHDF family (YTHDF1/2/3) and IGF2BP family (IGF2BP1/2/3), influence many biological processes by regulating the stability of downstream target mRNAs.[26] In recent years, the m6A modification of related genes mediated by the METTL3/IGF2BP1 axis has been extensively studied in a variety of human diseases.[27,28] A previous study suggested that the downregulation of METTL3 suppresses the oxidized low-density lipoprotein-induced Human Umbilical Vein Endothelial Cell proliferation, migration, and angiogenesis through IGF2BP1, thereby alleviating atherosclerosis progression.[29] Meanwhile, METTL3/IGF2BP1-mediated m6A modification promoted ferroptosis resistance in hepatoblastoma.[30] In TB, METTL3-mediated m6A modification restrained MTB-induced pyroptotic cell death in mouse macrophages,[16] confirming the positive role of METTL3 in TB progression. In the present work, we revealed that METTL3 was overexpressed, and the m6A level was upregulated in the MTB-induced macrophages. Further analysis confirmed that METTL3 mediated the m6A methylation of SMARCA5 to promote the latter’s mRNA stability, and IGF2BP1 directly recognized this modification. Rescue experiments verified that the si-METTL3-mediated inhibition of MTB-induced macrophage M1 polarization and inflammation could be abolished by SMARCA5 upregulation, confirming that METTL3 regulates SMARCA5 expression to facilitate MTB-induced macrophage functions and accelerate TB progression. This study has some limitations. Our research is currently limited to the cellular level. Owing to the lack of clinical information and samples, we are temporarily unable to analyze the correlation between SMARC5 expression and TB prognosis. In the future, we will consider collecting clinical samples and conducting animal experiments to further confirm the conclusions of this study.
Highlights
SMARCA5 knockdown inhibits MTB-induced macrophage M1 polarization and inflammation
METTL3/IGP2BP1 mediates the m6A methylation of SMARCA5
METTL3 enhances MTB-induced macrophage M1 polarization and inflammation by upregulating SMARCA5.
SUMMARYThe METTL3-mediated m6A methylation of SMARCA5 enhances MTB-induced macrophage M1 polarization and inflammation in an IGF2BP1-dependent manner. The proposed METTL3/SMARCA5 axis provides a potential molecular target for TB treatment, implying its clinical significance. Additional research is needed to confirm the METTL3/SMARCA5 axis as a TB treatment strategy.
Comments (0)