
Remember me
Glioblastoma, known for its rapid growth and invasive nature, is a formidable adversary in the realm of neuro-oncology, posing intricate challenges in the domains of therapeutic interventions and long-term prognostic outlooks.[1,2] In the complex tapestry of glioblastoma investigation, a disintegrin-like and metalloproteinase 15 (ADAM15) has emerged as a central orchestrator fueling the advancement and dissemination of tumors within the intricate microenvironment of the brain.[3,4] As a metallopeptidase enzyme, ADAM15 orchestrates a symphony of cellular processes, including adhesion, migration, and signaling, by cleaving diverse substrates.[5] Large amounts of available data indicate that ADAM15 plays a key role in diseases such as lung,[6] rectal,[7] and bladder cancer.[8] In addition, Xu et al. found that ADAM15 overexpression was associated with poor prognosis in hepatocellular carcinoma.[5] Simultaneously, protease-activated receptor 1 (PAR1), a pivotal G-protein-coupled receptor, exerts profound regulatory influences on the intricate processes of tumorigenesis and the relentless progression of diseases, thereby influencing cellular behavior and pathological development.[9] Previous reports have found that PAR1 is associated with microtubules and mitosis in glioma cells.[10] RAP1 was first discovered as a platelet thrombin receptor,[11] while protease-activated receptor 2 (PAR2) was not responsive to thrombin.[12]
At the cellular level, epithelial-mesenchymal transition (EMT) represents a highly complex and critical process characterized by the downregulation of epithelial markers and the upregulation of mesenchymal markers.[13,14] This molecular reprogramming facilitates the acquisition of migratory and invasive properties of cells, playing a crucial role in various physiological and pathological processes, such as embryogenesis, tissue regeneration, and cancer metastasis.[15,16] By investigating the intricate mechanisms underlying the crosstalk between ADAM15 and PAR1 in modulating the EMT pathway, the current research seeks to unveil the complex web of interactions that dictate the biological behavior of glioblastoma. Through a comprehensive analysis of how ADAM15 influences PAR1 activation within the framework of EMT, this study aspires to provide valuable insights into potential targets for therapeutic intervention and pharmaceutical innovations in the field of glioblastoma management, offering new perspectives for enhancing treatment strategies and patient outcomes.
MATERIAL AND METHODS Cells and reagentsThe U251 (Cat. No: BFN608006387) and U87 (Cat. No: BFN608006447) cell lines were procured from the American Type Culture Collection. These cell lines were cultured in Dulbecco’s modified eagle medium (DMEM) (Cat. No: 11965092, Gibco™, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Cat. No: A5670701, Gibco™, Grand Island, NY, USA) and penicillin-streptomycin (Cat. No:15140122, Gibco™, Grand Island, NY, USA) to support their growth and maintenance. The cells were maintained in a constant-temperature carbon dioxide (CO2) incubator at 37°C with 5% CO2. Before the experimentation, the cell lines were screened for mycoplasma contamination and validated for authenticity using short-tandem repeat (STR) analysis.
PAR1 antagonist: SCH79797 (Cat. No: SML1939, Merck Millipore, Billerica, MA, USA). PAR2 antagonist: FSLLRYNH2 Cat. No: 4751, R&D Systems Minneapolis, MN, USA). Restriction endonucleases (Cat. No: 3159, Takara, Tokyo, Japan) comprising HindIII, EcoRI, SacI, and XhoI were employed for DNA manipulation, while the T4 DNA ligase (Cat. No: 2011A, Takara, Tokyo, Japan) facilitated the ligation of DNA fragments. Furthermore, the SYBR Premix Ex TaqTM II (Cat. No: RR390Q, Takara, Tokyo, Japan) and reverse transcription reagents, specifically the PrimeScript™ RT reagent kit (Cat. No: RR037Q, Takara, Tokyo, Japan) were used for polymerase chain reaction (PCR) and reverse transcription experiments.
Cell transfectionThe U251 and U87 cells were divided into the ADAM15 overexpression group and the control group. The transfection reagent Lipofectamine 2000 was obtained from Invitrogen (Cat. No: 18501, Carlsbad, CA, USA). PEnCMV-ADAM15 plasmid was designed into ADAM15 overexpression and OE-NC. The ADAM15 overexpression sequence was synthesized by GeneChem (China, Shanghai). The transfection method was a slightly modified version of the one proposed by Xu et al.[5] The sense strand was 5’- AATTCAGCTTCCCTGGCTCTAGGCGAGTCGA CATATTGGCCTA-3’, and the antisense strand was 5’-CTAACTTGTCGACTCTCCAGAACGGTGTG CTTTGTGAG-3’. Plasmid extraction and gel recovery kits (Cat. No: 12123, QIAGEN, Dusseldorf, Germany). The si-NC was synthesized by GeneChem (China, Shanghai). The sense strand was 5’- AGCTTCCCTGG CTCTACCATACTGA-3’, and the antisense strand was 5’-CTAACTTGTCGACTTTGTGAG-3’.
The main steps were as follows: Synthesized RNA was polyadenylated further using poly(A) polymerase (Cat. No: 0AS72, American Research Products, Grandville, MI, USA) and dissolved in 150 mM potassium chloride at a final concentration of ∼100 ng/μL. Diluted RNA was filtered, heated at 90°C for 1 min, and then cooled on ice. Afterward, the transfection vector was mixed with the transfection reagents, and the mixture was then transfected into the U251 and U87 cells. After transfection, we continued to culture the cells to facilitate the successful entry of the transfection vectors into the cells and the expression of the genes or RNA contained within them. Finally, the transfection efficiency or expression of the transfection vectors was detected using qualitative real-time polymerase chain reaction (qRT-PCR) and western blot.
Transwell assayTranswell chambers (Cat. No: 3401)) and Matrigel-coated invasion membranes (Cat. No: 354480) were purchased from Corning Incorporated (NY, USA). The U251 and U87 cells were placed in a serum-free culture medium for 24 h of starvation treatment. At the same time, the matrix gel was placed in the refrigerator overnight to thaw and then cooled at 4°C. Then, the matrix gel was diluted to 1 mg/mL on ice, and 60 μL of the mixed solution was added to the transwell chamber and evenly spread on the bottom. After incubating at 37°C for 1–3 h, we carefully aspirated the unbound matrix gel. Then, serum-free medium was added, and the culture plate was placed in a 37°C incubator for hydration. Cell density was adjusted based on the number of cells seeded in the upper chamber. Then, the 24-well plate was incubated at 37°C, 5% CO2, and 90% humidity for 24 h. Finally, we fixed the cells for 30 min, which were stained by crystal violet (Cat. No: C0121, Beyotime, Shanghai, China) for 5 min, and then observed through a microscope (Cat. No: CX23, Olympus, Tokyo, Japan).
Clonogenic assayThe U251 and U87 cells were plated in a 24-well plate, cultured for 48 h, fixed by 4% paraformaldehyde (Cat. No: P0099, Beyotime, Shanghai, China), stained by crystal violet (Cat. No: C0121, Beyotime, Shanghai, China) for 10 min, and then observed through microscope (Cat. No: CX23, Olympus, Tokyo, Japan) to observe clonal formation.
EdU incorporationThe EdU incorporation kit (Cat. No: C2081, Beyotime, Beijing, China) was used to detect glioblastoma proliferation in the U251 and U87 cells. The cells were stained with EdU working solution, washed with phosphate-buffered saline (PBS), and then stained again with DAPI (Cat. No: D1306, Invitrogen, Carlsbad, CA, USA) to label the cell nuclei. After fixing the cells, they were air-dried at room temperature and observed under a fluorescence microscope (Cat. No: CX41-32RFL, Olympus, Tokyo, Japan) to assess proliferation in the U251 and U87 cells. The EdU-positive cells were marked by green fluorescence, while the cell nuclei were marked by blue fluorescence. The ratio of EdU-positive cells to total cells was recorded as the proliferative cell ratio.
Cell scratch assayThe cells were digested into single-cell suspension using trypsin and inoculated on a 6-well plate. Cells were cultured at 37 ℃ with 5% CO2 for 24 h. The next day, a 200 μL pipette head straight scratch was applied. PBS was used for washing three times. Finally, photographs are taken, and images are analyzed using ImageJ (v1.3.4, National Institutes of Health, Bethesda, MD, USA).
Western blotThe collected cells were mixed well with a radioimmunoprecipitation assay (Cat No. P0013, Beyotime, Shanghai, China). After the supernatant was collected, the protein concentrations were measured using the BCA kit (A045-4, Nanjing Institute of Bioengineering, Jiangsu, China). The sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel preparation kits (Cat. No: P0012AC, Beyotime, Beijing, China) were used in the protein analysis experiments. Total proteins were separated by SDS-PAGE gel electrophoresis and then subsequently transferred onto a polyvinylidene fluoride membrane (Cat. No: 08321S, Merck Millipore, Billerica, MA, USA). The membrane was then blocked with 5% skim milk at 37°C for 2 h. Primary antibodies ADAM15 (ab124698, 1:400), PAR1 (ab233741, 1:400), E-cadherin (ab40772, 1:400), N-cadherin (ab76011, 1:400), Vimentin (ab92547, 1:400), and glyceraldehyde-3-phosphate dehydrogenase (ab9485, 1:1000) were purchased from Abcam (Cambridge, MA, UK). Then, the antibodies were incubated with the membrane overnight at 4℃. Subsequently, the corresponding horseradish peroxidase (HRP) Anti-Rabbit immunoglobulin G (IgG) antibodies (Cat. No: ab288151, Abcam, Cambridge, MA, USA) were incubated with the membrane at 37°C for 1 h. Protein bands on the membrane were visualized using ECL luminescent fluid (BL520b, Biosharp Life Science, Hefei, Anhui, China) and then imaged using the Bio-Rad system (Cat. No: 12011319, Bio-Rad, Hercules, CA, USA). Finally, ImageJ (v 1.34, National Institutes of Health, Bethesda, MD, USA) was used to analyze the gray value of the strip.
qRT-PCRFollowing RNA precipitation, washing, dissolution, and concentration determination, total RNA was extracted from each experimental group post-transfection. The RNA extraction reagent Trizol (Cat. No: 15596018CN, Invitrogen, Carlsbad, CA, USA) was used in the RNA extraction processes. Reverse transcription of RNA into complementary DNA (cDNA) was conducted in accordance with the manufacturer’s instructions (Hifair® AdvanceFast 1st Strand cDNA Synthesis Kit, Cat. No: 11150, Yeasen, Shanghai, China). The resulting reaction solution was utilized for fluorescence quantitative PCR. The primer sequences used for the qPCR analysis are listed in Table 1. In this study, qRTPCR was performed in triplicates in multiple wells, with β-Actin as the internal reference. The 2-△△Ct method was employed for the results analysis.
Table 1: Primer sequences.
Primer Forward sequence (5’-3’) Reverse sequence (5’-3’) β-Actin TGTTGGCGTACAGGTCTTTGC GCTACGAGCTGCCTGACGG ADAM15 CCGCCGCTGCCAAATATAG GGCCTCAGGTAAACCAGTCTG PAR1 GCTGCCATCTCTACTTCCATC AAGCGGCGGTTGACATAGATT Immunofluorescence stainingFor the immunofluorescence staining, we followed the method of Wang and Liu,[17,18] with slight modification. The cells were fixed with 4% paraformaldehyde (Cat. No: P0099, Beyotime, Shanghai, China) at room temperature for 20 min. Permeabilization was conducted by adding 0.1% Triton X (Beyotime, P0096, Shanghai, China) for 10 min. Subsequently, blocking was performed for 1 h using 5% bovine serum albumin (Beyotime, ST023, Shanghai, China). Primary antibodies (Abcam, Cambridge, MA, USA) E-cadherin (ab40772, 1:400), Vimentin (ab8069, 1:400), and Ki67 primary antibody (Cat. No: ab16667, 1:1000) were then added and incubated overnight at 4 ℃. Following PBS rinsing, secondary antibodies Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) (1: 1000, ab150081, Abcam, Cambridge, MA, USA) and Goat Anti-Mouse IgG H&L (Alexa Fluor® 647) (1: 1000, ab150115, Abcam, Cambridge, MA, USA) were applied and incubated in darkness for 1 h at room temperature. This step was followed by treatment with DAPI (Cat. No: D1306, Invitrogen, Carlsbad, CA, USA) for 10 min. PBS was performed before sealing the slides. Fluorescence intensity analysis was conducted using a microscope (Olympus, BX63, Tokyo, Japan) and Image Pro Plus software (Version 6.0; Media Cybernetics, Silver Springs, MD, USA).
Statistical analysisWe followed the statistical analysis method of Wang et al.,[19] and GraphPad Prism (Version 8.0; La Jolla, CA, USA) was used for the statistical analysis and graphical representation of the results. Measurement data were presented as mean ± standard deviation. Comparison between two groups was performed using a t-test, while one-way analysis of variance (ANOVA) was used for comparisons involving multiple groups. The Tukey method was employed for multiple post hoc testing in one-way ANOVA. A significance level of P < 0.05 was considered statistically significant.
RESULTS Impacts of ADAM15 overexpression on tumor migration, invasion, and proliferationThe experimental groups consisted of cells overexpressing ADAM15, while the control groups consisted of cells without ADAM15 overexpression. Western blotting analysis was performed to confirm the efficiency of ADAM15 overexpression. The protein and messenger RNA (mRNA) levels of ADAM15 in the experimental (ADAM15 OE) groups were significantly increased compared to those in the control groups (P < 0.001) [Figure 1a-c]. Next, a scratch assay was conducted to evaluate the influence of ADAM15 on tumor migration. A defined scratch was made on the cell monolayer, and the closure of the scratch was observed and quantified. We observed that the relative migration rates were significantly increased in the experimental (ADAM15 OE) groups (P < 0.001) [Figure 1d-e].

Export to PPT
Meanwhile, a transwell chamber assay was performed to validate the migration of the U251 and U87 cells. We observed that the relative migration rates were significantly increased in the experimental (ADAM15 OE) groups in the U251 and U87 cells (P < 0.001) [Figure 1f and g]. As for the tumor invasion-promoting effects of ADAM15, a transwell chamber assay was performed using Matrigel-coated inserts. The results showed that the invasive abilities of ADAM15 overexpression groups in the U251 and U87 cells were significantly increased (P < 0.001) [Figure 1h and i]. These results demonstrated that ADAM15 overexpression significantly enhanced tumor migration and invasion, suggesting the role of ADAM15 in promoting the metastatic potential of glioblastoma cells.
Impacts of ADAM15 overexpression on the proliferation ability of glioblastoma cellsTo assess cell proliferation, clonogenic assays were performed. U251 and U87 cells from the experimental (ADAM15 OE) and control groups were plated at low density and allowed to form colonies. The results showed that the number and size of colonies in the ADAM15 OE group were significantly increased (P < 0.001) [Figure 2a and b]. Meanwhile, EdU incorporation was performed to detect the actively proliferating cells. The cells were exposed to EdU, which was incorporated into actively dividing cells during DNA synthesis. The levels of EdU incorporation in the ADAM15 OE group were significantly increased (P < 0.001) [Figure 2c and d]. As a marker of cell proliferation, immunohistochemistry staining using Ki67 antibody was conducted to determine the proportion of cells in the proliferative phase. The Ki67-positive cells were quantified, and the results showed that the ADAM15 OE group had more Ki67-positive cells (P < 0.001) [Figure 2e and f].
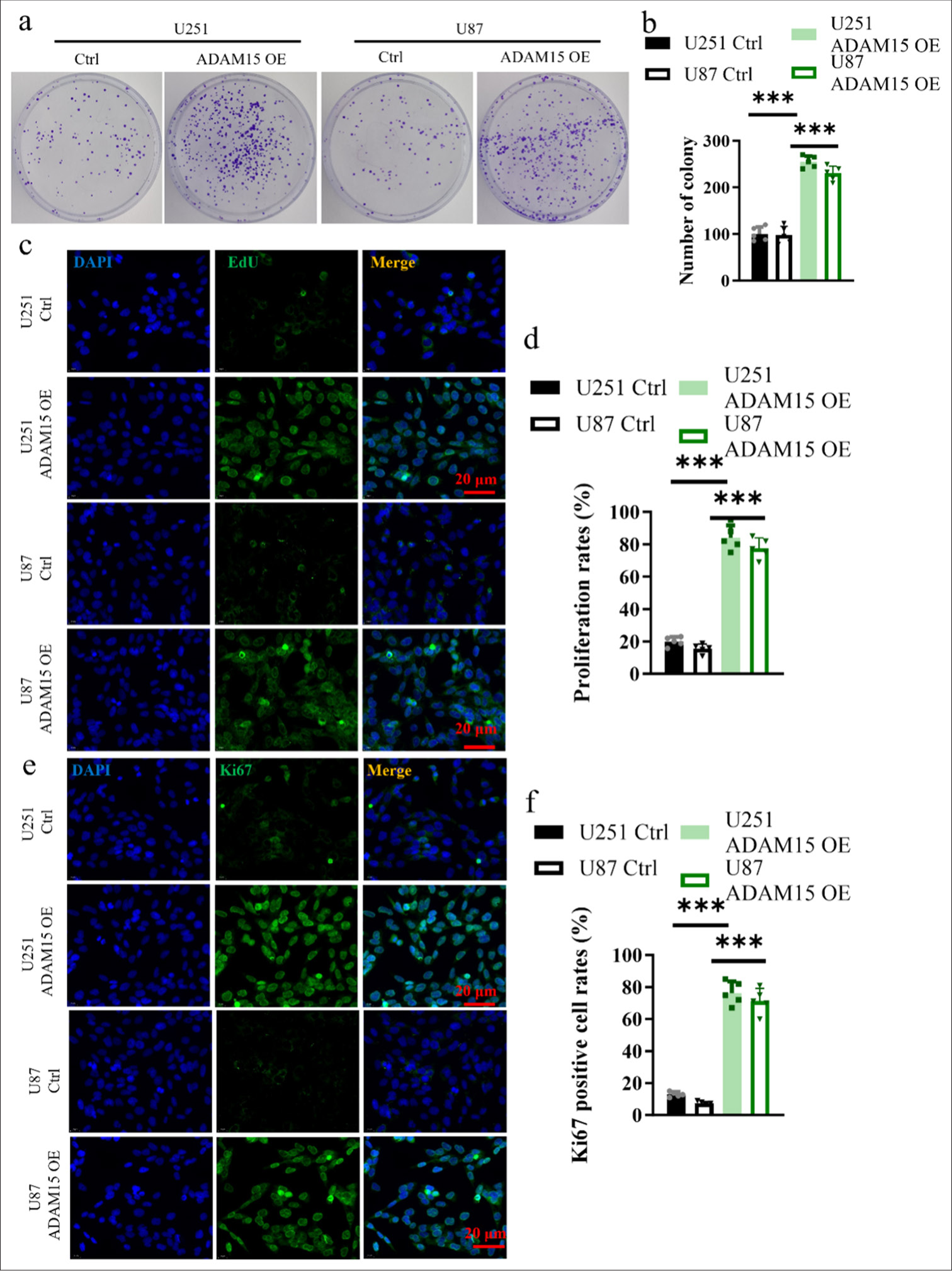
Export to PPT
The results conclusively showed that ADAM15 overexpression significantly enhanced the proliferation ability of glioblastoma cells. This finding strongly suggests that ADAM15 plays a crucial role in promoting glioblastoma cell proliferation.
Impacts of ADAM15 overexpression on PAR1 expression and EMTPAR1 is one of the four members of the protease-activated receptor family. PAR1 activation occurs when a protease, such as thrombin or matrix metalloproteinases, cleaves and exposes a specific peptide sequence on the receptor’s N-terminus.[20] This action leads to the activation of downstream signaling pathways that regulate EMT.
Western Blot and qRT-PCR analyses were performed to assess PAR1 expression, and the results showed that protein and mRNA levels of PAR1 in the experimental groups were significantly increased (P < 0.001) [Figure 3a-c]. The protein levels of N-cadherin and Vimentin in the experimental groups were significantly increased (P < 0.001) [Figure 3d-g].
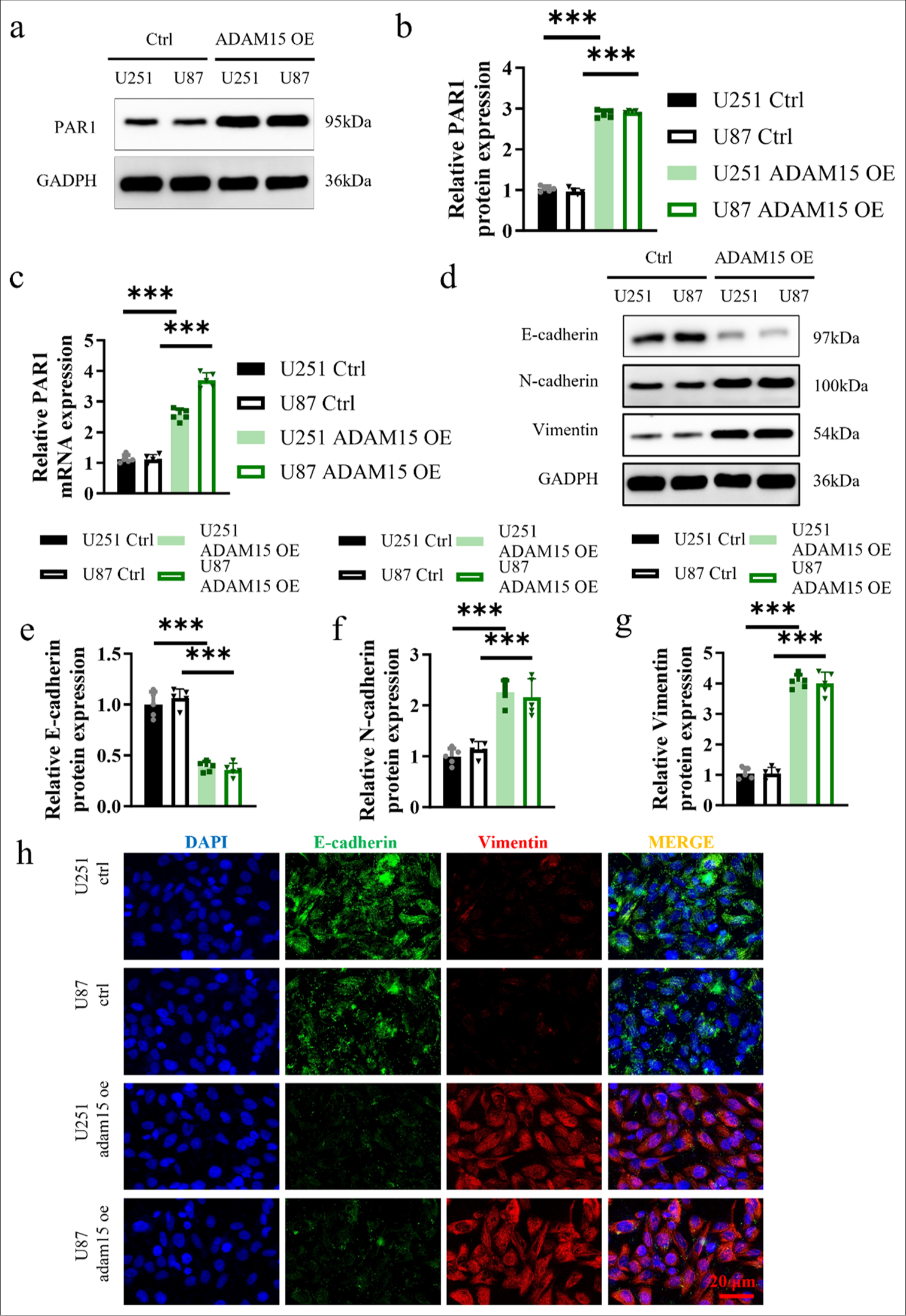
Export to PPT
Next, immunofluorescence staining of EMT markers was performed on the U251 and U87 cells from the ADAM15 OE and control groups to examine the localization and expression patterns of these markers. The results revealed a significantly higher co-staining ratio of EMT markers in the ADAM15 OE group [Figure 3h]. The result in Figure 3h suggests that the overexpression of ADAM15 may enhance EMT and upregulate the expression of EMT-associated proteins. Therefore, such findings provide further support for the role of ADAM15 in modulating the malignant characteristics of glioblastoma cells.
xInhibition of ADAM15-induced proliferation, migration, and invasion by PAR-1 antagonist treatmentThe U251 and U87 cells from the experimental group overexpressing ADAM15 were treated with either the PAR-1 antagonist SCH79797 or the PAR-2 antagonist FSLLRYNH2. We employed scratch assays to examine cell migration [Figures 4a and b], transwell assays to assess cell invasion [Figures 4c and d], and clonogenic formation assays to evaluate cell proliferation [Figures 4e and f]. The results showed that the administration of the PAR-1 antagonist SCH79797 significantly suppressed cell proliferation (P < 0.001), migration (P < 0.001), and invasion (P < 0.001) in the ADAM15 overexpression group. This finding suggests that the activation of PAR-1 may play a crucial role in mediating the effects of ADAM15 overexpression on promoting cell growth, movement, and invasion. Conversely, treatment with the PAR-2 antagonist FSLLRY-NH2 showed no significant inhibitory effect on cell proliferation (P = 0.87), migration (P = 0.93), or invasion (P = 0.59) in the experimental group. Thus, PAR-2 signaling may not be involved in the cellular processes influenced by ADAM15 overexpression.
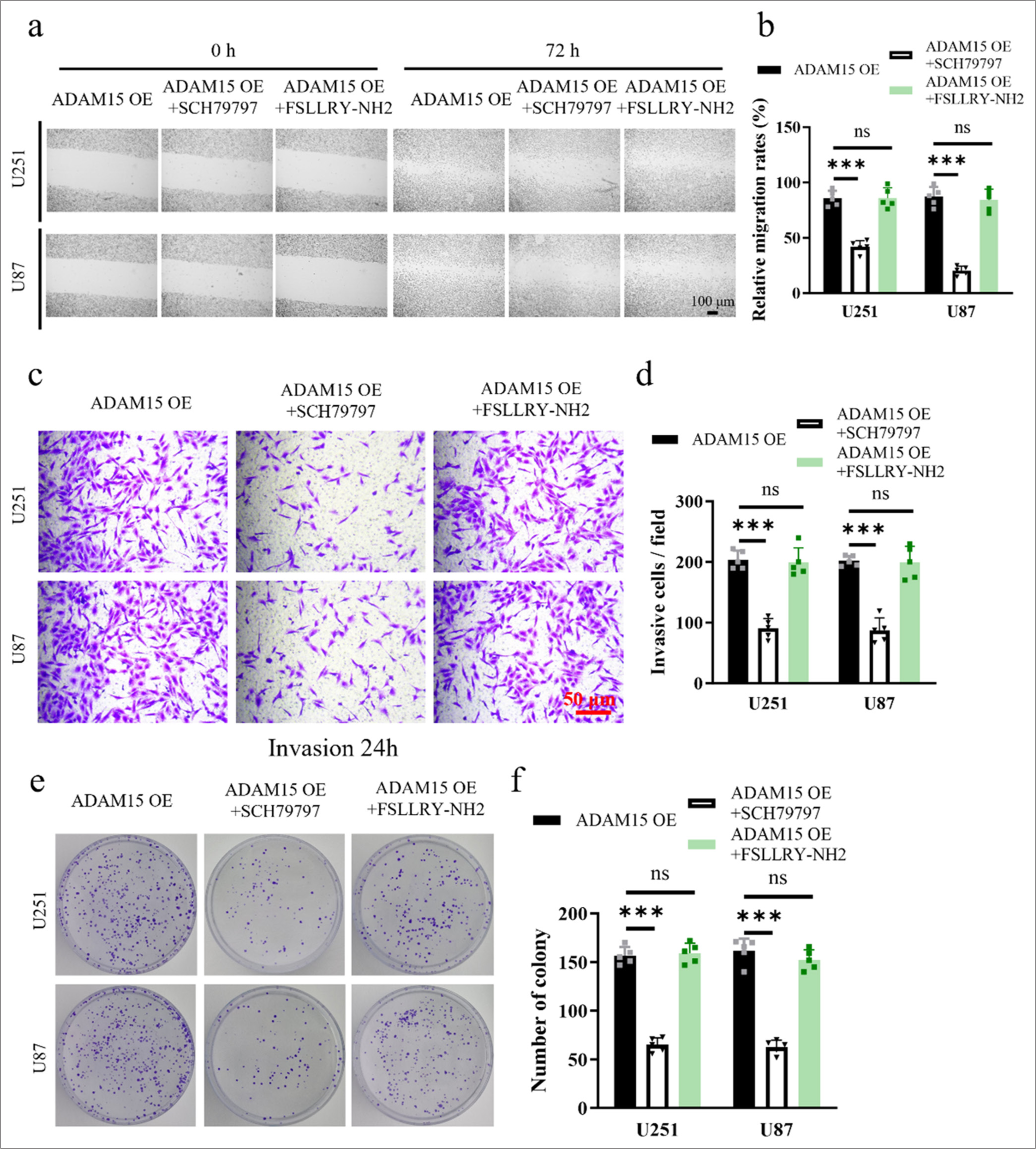
Export to PPT
In summary, these results provided deeper insights into the molecular mechanisms underlying the influence of ADAM15 on glioblastoma cell behaviors and highlighted the potential therapeutic implications of targeting PAR-1 for inhibiting ADAM15-driven tumor progression.
Inhibition of ADAM15-induced EMT by PAR-1 antagonist treatmentTo investigate the effects of PAR-1 antagonist treatment on EMT, the expression levels of EMT markers, such as E-cadherin, N-cadherin, and Vimentin, were analyzed using Western blot to assess EMT changes in U251 [Figure 5a and b] and U87 cells [Figure 5c and d]. Furthermore, immunofluorescence staining was performed on U251 and U87 cells from the ADAM15 OE group to examine the localization and expression levels of PAR-1 and EMT markers [Figure 5e]. On the one hand, treatment with the PAR-1 antagonist SCH79797 inhibited EMT in the cells, leading to a reduction in the protein expression (P < 0.01) and co-staining ratio of EMT markers in the SCH79797-treated group. This finding indicates that the overexpression of ADAM15 may promote epithelial-mesenchymal transition and decrease the expression of EMT-associated proteins.
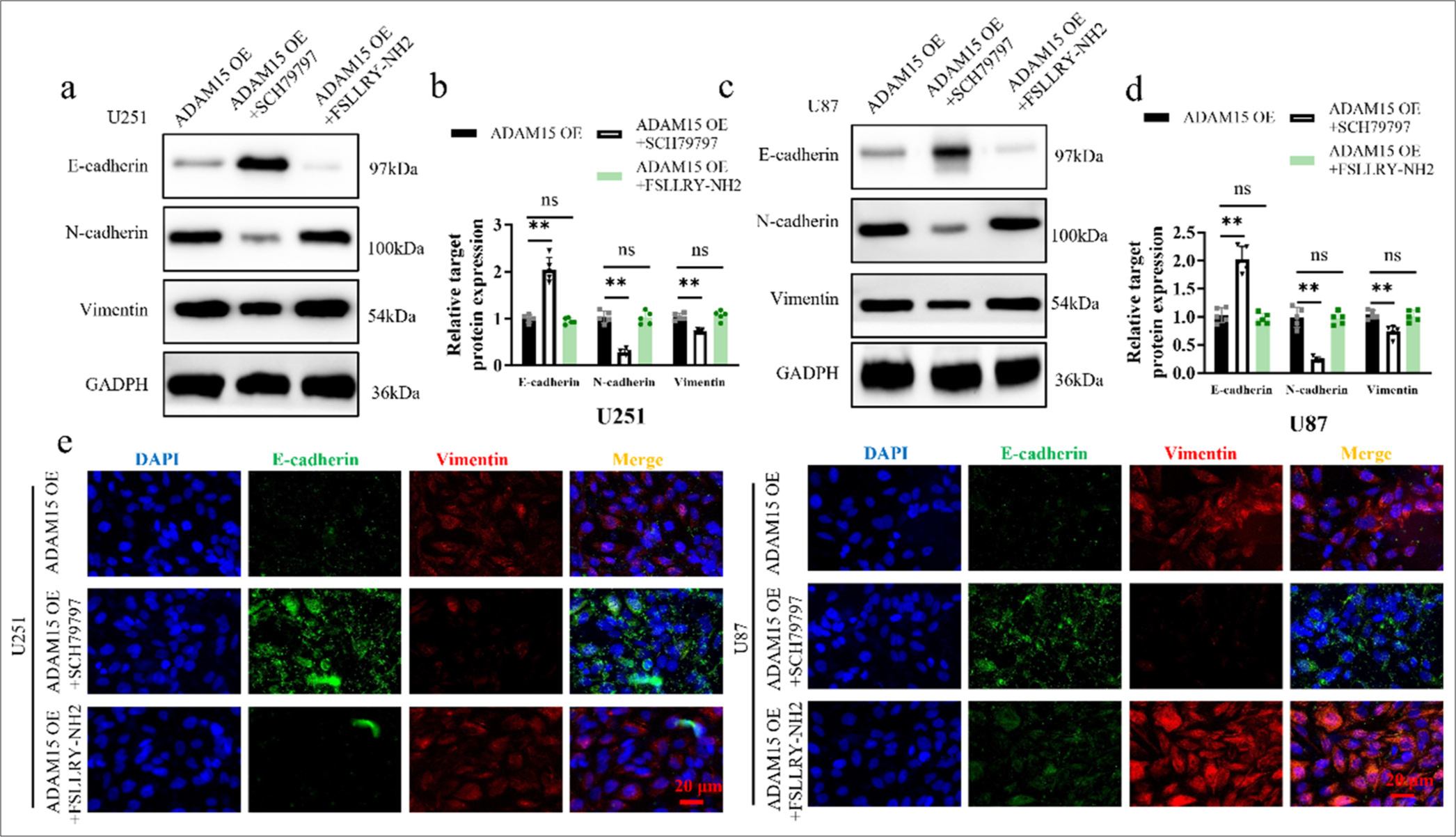
Export to PPT
On the other hand, the PAR-2 antagonist FSLLRY-NH2 had no inhibitory effect on EMT in the cells, suggesting that PAR-2 signaling may not be involved in the modulation of EMT driven by ADAM15 overexpression. These results not only shed light on the role of PAR-1 in mediating the EMT-promoting effects of ADAM15 but also highlight the potential of targeting PAR-1 as a strategy to inhibit ADAM15-induced EMT in glioblastoma cells.
DISCUSSIONIn this study, we investigated the intricate mechanisms through which ADAM15 influences glioblastoma proliferation and metastasis, focusing on its crucial role in activating the EMT pathway associated with PAR1. Through a meticulously designed series of experiments and rigorous data analysis, we demonstrated that the overexpression of ADAM15 significantly enhanced the proliferative and migratory capacities of glioblastoma cells, concomitant with pronounced changes in EMT-related markers. Notably, treatment with PAR1 inhibitors effectively attenuated the proliferation and migration induced by ADAM15 overexpression. This finding corroborates the notion that ADAM15 drives glioblastoma aggressiveness through the activation of the PAR1 signaling pathway.
ADAM15, also known as metargidin, is a multifunctional membrane-anchored metalloproteinase that plays a role in various cellular processes, including cell adhesion, migration, and proteolytic processing of cell surface proteins.[21] ADAM15 has been implicated in diverse physiological and pathological conditions, such as cancer progression,[21] inflammation,[22] and tissue remodeling.[23] Its ability to cleave and release extracellular domain-containing proteins from the cell surface makes it an important regulator of cell signaling and communication.[24]
PAR1 activation occurs through the cleavage of its N-terminus by various proteases, thus leading to the exposure of a tethered ligand that binds and activates the receptor.[20] This unique mechanism allows PAR1 to convert proteolytic cleavage events into intracellular signaling cascades, thus mediating cellular responses to a variety of stimuli.
Studies have shown that ADAM15 can cleave PAR1 at specific sites, resulting in the release of the tethered ligand and the activation of PAR1 signaling.[25] This shedding process is known to be important in regulating PAR1-mediated functions, such as cell migration, adhesion, and inflammatory responses. Furthermore, the dysregulation of ADAM15 and PAR1 signaling has been implicated in various diseases, including cancer metastasis, cardiovascular disorders, and inflammatory conditions.[26] Thus, understanding the complex interplay between ADAM15 and PAR1 sheds light on the potential therapeutic opportunities for targeting these molecules in the treatment of related pathologies.
Our study sheds new light on the critical involvement of ADAM15 in glioblastoma development, thereby offering a fresh perspective on potential therapeutic targets for combating this aggressive brain tumor. By modulating the EMT pathway, ADAM15 emerges as a promising target for future glioblastoma therapies. However, further investigations are warranted to unravel the intricate molecular mechanisms governing the crosstalk between ADAM15 and PAR1, paving the way for a more in-depth understanding of the molecular regulatory networks dictating glioblastoma progression.
Building upon our findings, related studies have underscored the significance of the ADAM protein family in driving cancer progression and metastasis.[27,28] For instance, the research conducted by Dutta et al. highlighted the pivotal role of ADAM proteins in promoting tumor invasion through EMT modulation in breast cancer cells.[29] Similarly, the work by Lu et al. demonstrated the regulatory function of ADAM15 in orchestrating metastatic processes in colorectal cancer by influencing the expression of EMT markers.[30,31]
The abovementioned studies collectively reinforce our findings and accentuate the multifaceted role of ADAM proteins in fueling cancer aggression through EMT-associated mechanisms. The integration of insights from these studies with our current research delineates a more holistic understanding of the complex signaling cascades governed by ADAM proteins in propelling cancer progression. Further advancements in research aimed at unraveling the intricate interplay between ADAM proteins, EMT pathways, and cancer progression hold significant promise in advancing therapeutic strategies for combating aggressive malignancies.
The study also has a limitation. In particular, animal experiments or patient-derived brain glioma tissue were not used in this study for more comprehensive exploration and discussion. Therefore, in the follow-up study, we will expand the sample size and enrich the sample types to further explore the role of ADAM15 in brain glioma.
SUMMARYOur research demonstrates that ADAM15 facilitates glioblastoma cell line proliferation and metastasis through the activation of the PAR1 and EMT, providing critical insights into the underlying mechanisms of glioblastoma pathogenesis. With the goal of providing theoretical basis and experimental support for innovative therapeutic strategies for glioblastoma treatment, in future studies, we will take the interaction between ADAM15 and PAR1 and their complex relationship with EMT pathway as our main research direction.
DATA AVAILABILITYData to support the findings of this study are available on reasonable request from the corresponding author.
ABBREVIATIONSADAM15: A disintegrin-like and metalloproteinase 15
DMEM: Dulbecco’s modified eagle medium
EMT: Epithelial-mesenchymal transition
PAR1: Protease-activated receptor 1
PAR2: Protease-activated receptor 2
qRT-PCR: Qualitative real-time polymerase chain reaction
STR: Short-tandem repeat
AUTHOR CONTRIBUTIONSRR and ZW.L: Designed the research study; ZW.L and QF: Performed the research; RR and QF: Collected and analyzed the data. All authors have been involved in drafting the manuscript and all authors have been involved in revising it critically for important intellectual content. All authors give final approval of the version to be published. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
Comments (0)