
Remember me
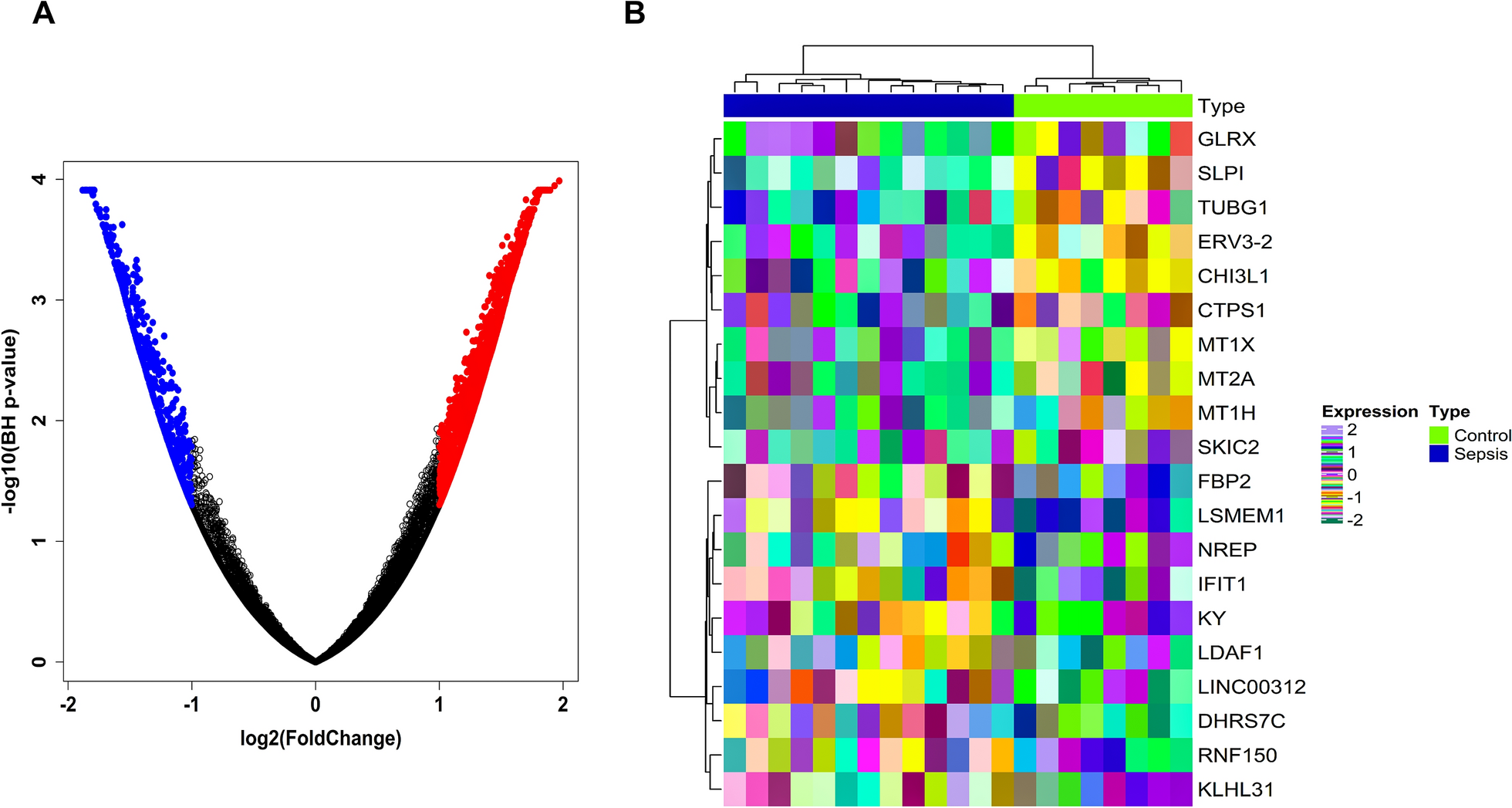





A pharmacophore is not only a set of common steric features, but also a set of electronic chemical features that show how a compound works at the active site of a specific biological macromolecule. Pharmacophore characteristics have been shown to be useful in observing ligand–protein interactions and in screening huge chemical databases for novel scaffolds that might serve as lead small molecules. The PDB entry 1s3b in association with chemicals was used to identify the lead drug against prostate cancer, and a pharmacophore model was then constructed. A small-molecule medication candidate based on the previously-identified antagonist need not be chemically synthesized; a compound library can be purchased instead. Twenty chemically synthesized active antagonists of the Monoamine oxidase B (MAOB) protein were identified using the well-known databases ChEMBL and an extensive literature search (Additional file 1: Table S1). PyRx performed molecular docking, and the antagonist CHEMBL3938629 had the highest binding score (PubChem CID: 56,961,657). The most significant binding was − 8.7 kcal/mol (Table 1). The overall schematic diagram for our research work has been provided by the flow chart (Fig. 1).
Table 1 List of 10 known active antagonist of monoamine oxidase B (MAOB) protein and their binding affinity towards the protein generated through molecular docking methodFig. 1
The overall schematic diagram for our research work has been provided by the flow chart (Fig. 1)
It is an essential to determine 3D structure of protein in the drug design and need to validate protein structure from the various protein data bank. The desire crystal X-ray structure of protein MAOB (PDB: 1S3B) was documented against antagonist and a structure-based pharmacophore model to the enzymatic cavity was generated. The ligands interaction with MAOB protein were determined experimentally and validated through X-ray diffraction method. The active site of MAOB protein have regulated by the binding of the inhibitor from the overall expression. The reliability of protein interaction with inhibitor ensured by the proper binding efficacy. Therefore, the active sites of inhibitor determined or examined to observe the sufficient interaction for getting more biological activity compared to the existing one. The key chemical features generated through the LigandScout 4.4 essential advance molecular design software based on pharmacophore model (Fig. 2).
Fig. 2
A The X-ray crystal structure of MAOB protein (PDB ID: 1s3b) was used to generate a 3D structure-based pharmacophore model of MAOB protein in combination with 46,781,908 (CID) ligands. B A number of pharmacophore features were generated following complex interaction; these include four yellow spheres, representing hydrophobic interaction; a shaded pink star shape, representing the positive ignitable with tolerance 2; three red arrows and spherical shapes, representing H bond acceptors with tolerance 1.5; and five hydrogen bond donors, represented by a green spherical or arrow shape
Fourteen distinct chemical characteristics were identified. As an example of a protein–ligand complex interaction, we showed that there were 43 characteristics, of which 33 were hydrophobic, 2 were positively ionizable bonds, 23 were H bond acceptors, 8 were H bond donors, and 42 had an exclusion volume (Fig. 2). Certain aspects have been left out of the pharmacophore model development process to ensure that the pharmacophore's optimal features are preserved.
The pharmacophore characteristics exhibited were formed by the protein–ligand interaction. The hydrophobic interactions generated with the amino acid residues of the chosen protein are prominent. There are several hydrogen bond donor interacted with protein whereas nitrogen atom in the benzene ring interact with HOH622A, HOH68OA, THR426A, HOH609A, SER15A, ARG42A and HOH607A (Fig. 3).
Fig. 3
The 2D structure of our chosen MAOB protein showed interactions with amino acid residues and hydrophobic contacts, which were shown in yellow color. The most common aspects of ligand–protein interactions, hydrogen bond donors (HBDs), were illustrated in red color, while HBAs' interactions with the oxygen and nitrogen atoms in the benzene ring and its various side chains were shown in blue color. The form and location of the binding pocket, which is maintained by hydrogen atoms and a constrained region, are not shown in the figure
Pharmacophore model validationValidation is an important part for the authentication of pharmacophore analysis as well as the quality of molecular model. The structure-based pharmacophore model generated before database screening because of models are able to differences with active compounds from the decoy set. Twenty active known MAOB antagonists with correspondences 600 decoy compounds in supplementary file (Additional file 2: Table S2) validated by enhance Database of Useful Decoys (DUDe). The IC50 values were merged with the decoy compounds and initially compound screened was track to validated model. The AUC value and EF value were estimated through the receiver operating curve (ROC). It was expressed the performance of a classification model that can give an idea about degree of reparability whereas AUC was utilized to describe the summery of the model performance. The higher AUC value proved the better predictability and it ranges between 0 and 1. However, the model shows the 100% correct prediction regarding of AUC value. Moreover, the early enrichment factor (EF1%) was 100 with an excellent AUC that was proved, the model has ability to distinguish true actives from the decoy compounds (Fig. 4).
Fig. 4
The ability of the structure-based pharmacophore model to find both active and decoy compounds allowed for the creation of a receiver operating characteristic (ROC) curve. Twenty MAOB actives and six hundred inactive compounds were used to test the pharmacophore model
Dataset generation for pharmacophore-base screeningThe identification of best lead molecules was an important part through the dataset generation during screening process. In the study, ZINC dataset utilized for collecting of commercially available chemical compounds. The information supplied included molecular weight, chemical structure, physical and chemical characteristics, and biologically active macromolecules. The ZINC data collection contains over 230 million chemical substances in 3D format that are publicly accessible via the website and ready to dock. Other compound information was also gathered from the ZINC data collection, such as Ambinter, which serves as a natural compound database library. The virtual screening database based on pharmacophores was recorded, and the pharmacophore model developed for each active chemical was uploaded to ZINCPharmer. Initially, the hits were calculated using the ZINC database of "ZINC natural products and ZINC natural derivatives," which contained millions of drug-like small molecules, natural products, and FDA-approved medications. The expected values for the RMSD sphere center fed into ZINCPharmer were 0.5Å, and a total of 11,000 compounds were returned for further screening. Finally, the hit was counted and save as well as downloaded for further screening.
Pharmacophore-based virtual screeningThe protein–ligand complex structure has documented through the purchasable compounds library for the generation of pharmacophore feature. The relative pharmacophore has been used for scoring function during the screening process and omitted maximum four features for all query features. It is a quiet hard to query all features during the screening process, for this reasons, few features omitted to enhance the ability of pharmacophore fit score. The higher score can be sowed the better activity against the targeted macromolecules and fitted with the desire environment. As a result, the ROC curve displays the pharmacophore fit value for geometric feature fit to the 3D-structure-based pharmacophore model (Fig. 4). The protein with the highest match score to the verified pharmacophore model demonstrates action against our desired MAOB protein. As a result, the chemical was labeled a cornuted hit, recovered, and stored for future analysis.
Molecular docking based virtual screeningThe desired MAOB protein has two ligands attached to it (PDB ID: 1S3B). The protein pockets have several points of attachment and a variety of shapes for ligand binding. From this particular protein, the binding location of the complex structure's active site has been obtained and determined. The active site (AS) of the protein is formed by combining several AA residues in a specific area that has the ability to form a transient connection with the substrate, known as the binding site. The protein's active site serves as the chemical substrate for the reaction that is being catalyzed. The protein's binding site can identify ligands and form a strong binding connection with the protein in order to stabilize intermediate reactions. The CASTpi server was used to identify the MAOB protein active sites, and the combined binding positions of the active sites were then obtained. The protein's estimated active pocket helps to recover the binding site residue. Total twelve active sites identified from the MAOB protein and further confirmed through the PrankWeb (https://prankweb.cz/) server. The active site pocket was analyzed and binding site position at ASN117, ASP114, MET 125, GLU142, TRP 143, CYS156, LEU151, LYS149, SER488, VAL489, PRO490 and ARG494 that have been depict by the different color such as red, deep green, green, yellow, deep pink, pink, orange, deep blue, mild blue, blue, black, and white, (Fig. 5). The server-identified binding sites were used to build a receptor grid during the molecular docking simulation process, with grid box dimensions X = 98.39, Y = 102.46, and Z = 69.21 in angstrom (Å).
Fig. 5
The active site and corresponding binding site of the MAOB protein are shown. The active site and its corresponding aa are represented Ball shapes atom with red, black, orange, white, pink and blue colors, respectively
Molecular dockingAs a vital part of the drug discovery process, molecular docking is being used in this work to evaluate how well the hit compounds attach to the target MAOB protein. Using the assumptions of the pharmacophore model as a guide, the PyRx tools Autodock Vina were used to dock drugs onto MAOB and assess their binding affinity. Four compounds were identified to have higher binding affinities than the MAOB antagonist ZINC ID 8215434 (− 9.2 kcal/mol), which was employed in the primary pharmacophore model creation (ZINC ID 12143050, ZINC ID 08301324, ZINC ID 16743012, and ZINC ID 64165826; Table 2). All of the hit chemicals in Additional file 2: Table S2 have slightly different binding affinities. Interestingly, it was hypothesized that drugs with higher docking scores would bind the target protein more effectively.
Table 2 Docking score with MAOB protein, docking score and molecular weight of the top four selected CompoundsInterpretation of protein–ligands interactionsThe interpretation of protein–ligands interaction showed in the ZINC ID: 12,143,050 formed three conventional hydrogen bond interactions with TYR435, SER59 and TYR60, seven van der Waals interaction with PHE168, LEU328, LYS296, PHE343, GLN206, ARG42, GLY58, two pi carbon hydrogen bond with GLY434 and ILE199, two pi sigma CYS172 and ILE 198, one pi sulfur bond with CYS 397 and three Pi-alkyl bonds with TYR398, GLY57 and TYR326 with desire MAOB protein (Figs. 5A and 6A). In the case of ZINC08301324 van der walls bonds were predominantly formed with GLY11, GLU34, VAL10, PRO234, ILE272, LYS271, LEU268, ALA263, SER394, GLY13, SER15, THR43, MET436, THR426, TYR435, GLY434, TYR398, and CYS397. The position of VAL235, TYR393 and ARG42 acquired three conventional hydrogen bonds, two pi sigma bond ALA35 and ILE264 and THREE alkyl bond with PRO265, ILE14 and ALA439 position (Figs. 6B and 7B).
Fig. 6
The protein–ligand complexes interact in three dimensions. A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826 depict ligand interaction with the protein MOAB after molecular docking
Fig. 7
The protein–ligand complexes are able to interact in two dimensions. After molecular docking, the ligand makes contact with the MOAB protein, as shown in A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826. Colors like blue, red, purple, light pink, deep pink, green and sky blue were used to denote various forms of bonds
For the compound ZINC16743012, the number of van der Waals interaction at position TYR60, PHE343, CYS397, GLY97, THR43, GLY40, ILE14, GLY13, ALA263, SER15, GLY425, GLY434, SER59 and TYR435 has found to be formed. Three conventional hydrogen bonds with GLN206, THR426 and MET436 position, one unfavorable positive-positive bond and one pi alkyl bond at ALA 439 have found to be formed (Figs. 6C and 7C). On the other hand, ZINC64165826, it has observed to formed six van der Waals carbon-hydrogen bonding in the position of PRO104, PHE99, VAL92, THR111, SER 160, and TRP107, three hydrogen bond with TYR97, ASN108 and HIS90, one pi-Alkyl bond with at position VAL106 (Fig. 6D).
Absorption, distribution, metabolism and excretion (ADME) and toxicity test Analysis of ADME propertiesThe drug is absorbed, distributed, metabolized, and excreted after being administered to an animal model or a human, resulting in active or passive transport to the target site. A pharmacological interaction with a biological macromolecule can be either positive or negative. The design of a drug is a step-by-step process, and failing to do so will lead to it being rejected, which is expensive for the company. The bioavailability of a drug is determined by its safety and efficacy, and lack of safety and efficacy are the primary reasons for drug failure, which are determined primarily by its ADME properties. SwissADME was utilized to assess the ADME qualities of the four compounds in terms of their pharmacokinetic parameters, including lipophilicity, water solubility, drug-likeness, and medicinal chemistry. The lipophilicity of a substance indicates that it may easily diffuse across the cell membrane; hence, an oral preparation is inappropriate. Moreover, since gastrointestinal absorption is limited, an injectable dose form may be an efficient way for achieving a quick beginning of action (Table 3). In addition, CoMFA stands for Comparative Molecular Field Analysis, an extensively employed 3D-QSAR method. Creating a three-dimensional grid around a molecule of interest and calculating the steric (shape) and electrostatic properties at each grid point are required (Fig. 8).
Table 3 List of pharmacokinetic properties (physico-chemical, lipophilicity, water solubility, drug likeness, and medicinal chemistry) of the selected four compoundsFig. 8
Creating a three-dimensional grid around a molecule of interest and calculating the steric (shape) and electrostatic properties at each grid point through the (CoMFA) Comparative Molecular Field Analysis for four selected compounds
CoMFA result can provide valuable insights into the structure activity relationship of the studied molecules. This information can aid in the design of novel compounds with the desired activity profiles by assisting researchers in gaining a better understanding of the factors that influence the biological activity of the molecules. The spatial arrangement of the four finest compounds results in steric effects. Four compounds, including ZINC12143050, ZINC8301324, ZINC16743012, and ZINC64165826, yielded experimental activity values of 5.22184875, 5.21184875, 5.21184875, and 5.21184875, respectively (Additional file 3: Table S3).
Pharmacophore features analysisA pharmacophore is a collection of either steric determinants or electronic properties that confirms optimal supramolecular interactions when conducting virtual screenings on large databases of molecules. Molecular docking is a powerful and more efficient method of finding molecules against specific targets that can induce or inhibit macromolecular activity. The compound with similar or relevant properties should exhibit the same or better activity as the query compound. Based on the docking score analysis, the pharmacophore features of ZINC12143050, ZINC08301324, ZINC16743012, and ZINC64165826, along with the antagonist CHEMBL3938629, were analyzed and compared. All the compounds have better pharmacophore properties than the antagonist CID: 56,961,657, so they are expected to be effective against our target protein. The pharmacophore feature found of the four compounds has shown in Fig. 9.
Fig. 9
Pharmacophore features generated from the four selected compounds attach to the desire MAOB protein. Ligands attaching to A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826 have better pharmacophore features than antagonist CID: 56,961,657
The evaluation of in-silico toxicity is an essential step that must be completed before clinical trials can begin for the purpose of selecting more effective lead compounds. Because of their precision, speed, and accessibility, computer-based toxicity measures have gained a lot of popularity in recent years. These qualities allow them to offer information on any molecule, whether it be natural or manufactured. Both the no-cost TEST tool and the ProTox II server were put to use as part of our investigation into the potential dangers posed by the four substances that were chosen. A number of toxicological parameters, including as acute toxicity, hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity, and immunotoxicity, were examined by the various software packages. Based on these evaluations, a median lethal dosage (LD50) in mg/kg was derived. According to the ProTox-II service, compounds ZINC12143050, ZINC08301324, and ZINC64165826 were classified as belonging to class 4, and LD50 ranges have also been compiled (Table 4).
Table 4 List of toxicity properties (organ toxicity, toxicity endpoints, Tox21-nuclear receptor signaling pathways, Tox21-Stress response pathway, fathead minnow LC50 (96 h), developmental toxicity, oral rat LD50,bioaccumulation factor) of the selected compoundsMolecular dynamics (MD) simulationMD simulations are used to investigate the binding stability of protein–ligand docking complexes. As an added benefit, MD simulations reveal details regarding intermolecular interactions over a given time scale. Here we use MD simulation methods to examine the docking contacts of four natural compounds and one reference antagonist with the MAOB protein, checking the stability of the protein-molecule complex and the strength of its intermolecular connections in less than 100 ns. The Maestro Desmond interface was used for MD trajectory extraction with SID, and the RMSD, RMSF, and Protein–Ligand (P-L) interaction mapping statistics were used to display the simulation results.
RMSD analysisIn MD simulation, the root mean square deviation (RMSD) is used to calculate the average distance caused by atom displacement for a given time frame in comparison with a reference time frame. The RMSD value of specific protein structure such as Cα, backbone, sidechain and heavy atoms have been estimated. The RMSD of the protein fit ligand captured from all the time frames during the reference time (in our case 100 ns). The RMSD value calculated from the X frame. It can be established whether the simulation has equilibrated or not based on the RMSD result. Within a reference protein structure, fluctuations of 1–3 Å are perfectly acceptable, but much greater values are not. A substantial conformational alteration in the protein indicates that the system is unstable. Except in the ZINC12143050 combination, the Cα atoms of MAOB displayed acceptable fluctuations in our four protein–ligand docking complexes. During a 100 ns simulation experiment, the compound ZINC12143050 showed an extended variation of 5.1 Å and a maximum fluctuation of 8.81 (between 26 and 28 ns) (Fig. 10). According to the data, MAOB undergoes protein conformation changes as a result of ZINC12143050 binding. Furthermore, at the end of the 100 ns simulation interval, measurement of RMSD using data acquired from protein fit ligands revealed minimal fluctuations (4.84 Å).
Fig. 10
Showing the RMSD values of the (MAOB) in complex with the selected four compounds A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826 extracted from Cα atoms of the complex system
RMSF analysisThe local conformational change in the protein chain and the ligand molecules must be identified and quantified using the Root Mean Square Fluctuation (RMSF). When the MAOB protein was in contact with natural chemicals, the alterations caused by the residue index C were utilized to compute the local structural fluctuations. It's interesting to note that, with the exception of the N-terminal minimum 1.80 to maximum 9.90, all protein residues exhibit low RMSF values. With the exception of compound ZINC12143050, the combination of probable drugs against MOAB protein was validated by looking at the RMSF and RMSD values for all protein–ligand complexes. Hence, except for compound ZINC1070004335, the combined screened potential compounds were supported by examination of RMSF and RMSD values for all protein–ligand complexes. The apoprotein showed the most variation between residue positions 105 aa, with a fluctuation of 2.0 at PRO105. A minimal amount of variation was also visible in the apo structure at residue position THR241. The molecule Zinc 12,143,050 in combination with the protein was then compared to the apo structure, and a significant variation at residue position PRO333 was discovered (Fig. 11). In comparison with the apoprotein structure, zinc 08301324 appears to have the lowest average RMSF range between 1.0 and 1.3, and the variation of ASP153 and ALA355 was similarly minimal. The RMSF graph, on the other hand, revealed that the MOAB protein had lower average low and significant values in association with Zinc 16,743,012 (0.99 to 1.03) and Zinc 64,165,826 (1.3) than the reference apo structure. As previously mentioned, a low RMSF value denotes better protein stability, whereas the RMSF values discovered in this study for each protein–ligand system were lower than those for apoprotein. As a result, it is anticipated that the chemicals will maintain a stable interaction with the protein without changing its structure.
Fig. 11
Showing the RMSD values of the (MAOB) in complex with the selected four compounds A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826 extracted from Cα atoms of the complex system
Protein–ligand interaction analysisHydrogen bonding, ionic bonding, water bridges, and hydrophobic bonding all play a role in turning a molecule into an effective drug. The MAOB protein had protein–ligand contact, and four natural chemicals were chosen from the MD trajectories and studied using the Desmond module's default parameters. All of the natural chemicals tested, ZINC12143050, ZINC08301324, ZINC16743012, and ZINC64165826, had tangible contact with the majority of the protein residues (Fig. 12). Furthermore, the four compounds evaluated using different filtering methods showed significant intermolecular interaction.
Fig. 12
The interactions between proteins and ligands are showing for 100 ns. The interaction of selected four compounds A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826 in complex with the (MAOB)
Solvent accessible surface areaBiological macromolecule structure and function are both influenced by their solvent-accessible surface area (SASA). Protein surface amino acid residues operate as active sites and bind ligands, providing insight into the solvent-like behavior (hydrophilic or hydrophobic) of molecules and protein–ligand complexes. Therefore, Fig. 13 displays the computed SASA value of the protein in association with the chemicals (A) ZINC12143050, (B) ZINC08301324, (C) ZINC16743012, and (D) ZINC64165826. For complicated systems, an average SASA value of 0–40 A was found, which indicates a significant amount of exposure of an amino acid residue to the molecule of interest.
Fig. 13
Solvent accessible surface area (SASA) of the protein– ligand complex was calculated from the compounds Zinc ID: A ZINC12143050, B ZINC08301324, C ZINC16743012, and D ZINC64165826 until 100 ns simulation
Comments (0)