
Remember me
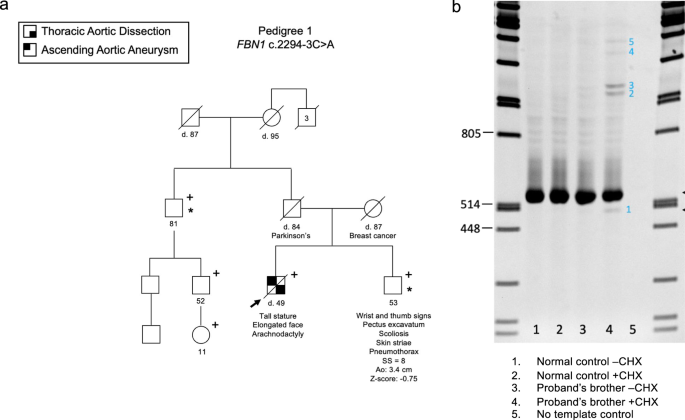
We obtained WGS from 1182 de-identified subjects with 22q11.2DS from the International 22q11.2 Brain and Behavior Consortium (IBBC)10,11,12,13. We then performed quality control measures and after this, there were a total of over 21 million variants that remained (Supplementary Fig. 1). The number of single nucleotide variants and indels with one or more alternative alleles were determined (Supplementary Fig. 2a). In this cohort, indels comprised 8.62% of the total number of variants (Supplementary Fig. 2a). Most of the indels were within 10 bp in size (Supplementary Fig. 2b). The alternate allele frequency distribution for the cohort versus the number of variants were identified and most of the variants occurred in only one subject (Supplementary Fig. 2c). Principal component analysis (PCA) was then performed to identify the ancestry groups among the 1182 subjects with 22q11.2DS (Supplementary Fig. 3; Supplementary Table 1). The subjects were distributed in Caucasian, African Descent and Hispanic ancestry groups, using the HapMap population (Supplementary Fig. 3a, b).
Study designTo identify genetic modifiers, we compared 456 cases with CTDs (22q11.2DS-CTDs) and 537 controls without CHD, all with 22q11.2DS (Fig. 1a; Supplementary Table 1). We excluded those with isolated ventricular septal or atrial septal defects because their developmental origins are more complex than for CTDs. We focused on rare variants that might affect the function of proteins along with haploinsufficiency of genes in the 22q11.2 region, during cardiac development. Our study design is illustrated in the flow diagram in Fig. 1b. We applied PEMapper/PECaller14 to identify variants and we then validated with GATK15 (Fig. 1b; Supplementary Fig. 4). The purpose of using the two different variant calling algorithms was to rule out false positive variants. Rare variants had an alternative allele frequency (AAF) in genomAD of less than 1% (Fig. 1b). Comprehensive annotation software and algorithms were used (see Methods) to identify 55,379 variants including LoF, splicing and putative damaging missense variants (Supplementary Table 2; Supplementary Fig. 4). Additional software and algorithms were used for further filtering (phastCons > = 0.5, for evolutionarily conserved variants; CADD > = 10 for deleteriousness of variants, CCRS > = 80 for constrained coding regions; Supplementary Fig. 4) to identify 1861 high-confidence, predicted most damaging rare coding/splicing variants (MDRV, Fig. 1b; Supplementary Table 3; Supplementary Figs. 4 and 5).
Fig. 1: The 22q11.2 deletion syndrome cohort and study design.
a Pie chart of intracardiac and aortic arch phenotypes. Control (gray, no significant heart defect); CTD (conotruncal heart defect, blue); ASD alone (isolated atrial septal defect but no other heart or aortic arch defects, light blue); VSD alone (isolated ventricular septal defect but no other heart or aortic arch defects, light blue). Pie chart includes controls (gray) versus CTD cases with phenotypes including: TOF (tetralogy of Fallot, light blue), RAA (right sided aortic arch, orange), IAAB (interrupted aortic arch type B, green), PTA (persistent truncus arteriosus, yellow), PS/PA (pulmonary stenosis and/or pulmonic atresia, blue) and other aortic arch defects such as abnormal origin of the right or left subclavian artery, alone (light green). b Schematic representation of the case-control study design using WGS. Variants were identified using PEMapper/Caller and validated by GATK. Only shared variants between both pipelines were used. Following quality control measures of the raw WGS data, variant annotation was performed to identify rare (< 1%) predicted LoF (loss of function), damaging splicing and damaging missense variants followed by filtering-based annotation on phastCons (conservation), CADD and CCRS (constrained coding regions) scores to identify MDRVs. Then gene set analyses were performed including over-representation (ORA) and weighted gene set based tests. STRING analysis was performed to identify potential biological network interactions. c Lateral side of mouse embryo at E10.5 with outline of tissues used for bulk RNA-sequencing (pharyngeal arches 2-6, PA2-6, blue; outflow tract and right ventricle, OFT + RV, green; left ventricle and atria, LV+atria, yellow).
Once the variants were identified, the number of MDRVs were calculated per subject. A total of 1.78 MDRVs were found per subject in 22q11.2DS-CTD cases and 1.85 MDRVs were found per subject in controls, with no significant overall excess of MDRVs in either group (P = 0.485, Supplementary Fig. 6a). Male and female subjects had similar burden of MDRVs (1.81 versus 1.79, P = 0.803). Stratified analysis demonstrated there was no significant difference of MDRV burden between 22q11.2DS-CTD cases and controls within each of three main ancestry groups (all P > 0.05, Supplementary Fig. 3a and Supplementary Table 4), indicating that cases and controls were well matched for ancestry. The identified 1861 MDRVs affected 1261 genes, of which 83% had just one MDRV (1048/1261; Supplementary Fig. 6b).
Tbx1 is expressed in the pharyngeal apparatus (PA; arches 2–6) and in cells that migrate to the cardiac OFT and aortic arch arteries between mouse embryonic day (E)8-10.516. To filter for genes most likely to be relevant to progenitor populations of the heart and TBX1, we generated RNA-seq data to determine the expression level of genes in the PA, OFT with right ventricle (OFT + RV), and/or left ventricle with atria (LV+atria) of mouse embryos at E10.5 (Fig. 1c). Of the protein coding genes in the mouse genome in each tissue, we considered the top 35% as expressed (with > = 25 reads per million; Supplementary Table 5) and referred to them as cardiac progenitor cells.
Chromatin regulatory genes identified with recurrent MDRVs in 22q11.2DS-CTD casesWe selected recurrently affected genes with MDRVs in at least two cases and no controls to help avoid possible random sequencing artifacts. A similar criterion was applied to the control group (two or more controls, no cases; Methods). In the 456, 22q11.2DS-CTD cases, there were 413 genes with MDRVs, 31 of which were recurrently affected. In 537 controls, there were 466 genes with MDRVs, and 38 were recurrently affected. We found 12 of 31 recurrent genes in cases (Table 1; 5.7% of cases), and 13 of 38 recurrent genes in controls, were expressed in mouse embryonic cardiac progenitors (Supplementary Table 5). In total, we did not find more genes affected in cases versus controls with 22q11.2DS. We next wanted to focus on the functional categories of genes identified in cases versus controls that were expressed in cardiac progenitor cells.
Table 1 Twelve recurrently affected cardiac developmental expressed genes with MDRVs identified in CTD cases (and none in controls) in individuals with 22q11.2DS.Strikingly, half of the 12 recurrent genes with expression in cardiac progenitor cells in 22q11.2DS-CTD cases were chromatin regulatory genes: NSD1, KAT6A, KMT2D, PHF21A, EP400 and KMT2C, of which some are associated with known syndromes that include CHD, supporting their candidacy (Table 1). Table 2 lists the detailed annotation of the MDRVs identified in the six recurrently affected chromatin regulatory genes, as well as the associated cardiac phenotypes. The most frequent cardiac phenotype was tetralogy of Fallot followed in frequency by right sided aortic arch (Table 2). Moreover, there were MDRVs in KMT2C in two additional 22q11.2DS participants (not included here as CTD cases or as controls), one with a ventricular septal defect (VSD; with no other heart or aortic arch anomalies) and one with an atrial septal defect (ASD; with no other heart or aortic arch anomalies; Tables 1 and 2). Although there was no increase in the presence of MDRVs, thus, no increase in burden, we found a specific enrichment of a particular functional class of genes. We found that chromatin regulatory genes were greatly enriched among all possible functional categories of genes in the genome, whereas no single functional category of genes was enriched in controls.
Table 2 Identified MDRVs in the six recurrently affected chromatin genes in CTD cases.Over-representation analysis of recurrent genes identifies chromatin regulatory genes in 22q11.2-CTD cases but not controlsConsidering both the rarity of high-confidence MDRVs and relatively limited number of samples included, we next used a gene set based approach to test the aggregated effect of MDRVs in multiple functionally or conceptually connected genes in 19 gene sets, in cases and controls (Fig. 1b). The 19 gene sets were derived from three different sources of which had between 100 and 1000 genes per set (Supplementary Table 6). There was a total of 5403 genes in the 19 gene sets, of which 347 had one or more MDRVs. This number of genes per set ensured sufficient power for our statistical tests. One source included constrained, haploinsufficiency, and essential gene sets that are under strong purifying selection17, tend to play important roles in protein interaction networks18,19, and are highly enriched for disease genes20. The second source was chromatin regulatory gene sets because some of this functional category was identified in this study (Table 1). Additionally, de novo mutations in chromatin regulatory genes were found in individuals with sporadic CHD by the Pediatric Cardiac Genomics Consortium (PCGC)21. One gene set consisted of 866 genes (All Chromatin Regulatory Genes, combined from several sources; Supplementary Table 7) and the other consisted of 274 genes and was a smaller subset (REACTOME term Chromatin Modifying Enzymes, Supplementary Tables 6 and 7). The third source comprised 14 gene sets (116 to 791 genes per set), derived from genetic studies of sporadic CHD by the PCGC, including genes found with mutations in one or more subjects7. These 19 gene sets, though curated using differing concepts, are not mutually exclusive, as shown in the Venn plots in Supplementary Fig. 7, indicating various degrees of overlap among different gene sets.
We then performed an over-representation analysis (ORA) of the recurrently affected genes on 19 gene sets (Fig. 2). We found that five of the six recurrent chromatin regulatory genes described above (Table 1; not EP400) contributed to findings for the Constrained Genes set with borderline significance (n = 968; Fig. 2, panel 1, P = 5.28 × 10−3). The chromatin gene sets showed significant over-representation, with all six genes shown in Table 2, either when filtered by expression in cardiac progenitor cells or not filtered (Fig. 2, panels 1 and 2; Supplementary Table 8). Further, four of these six genes contributed to findings for the Candidate CHD Genes set created by the PCGC (not EP400 or PHF21A; n = 402; Fig. 2, panel 2). None of the other gene sets were overrepresented nor were the three non-cardiac gene sets used as controls (Fig. 2; Supplementary Table 8). In the control 22q11.2DS samples, there was no enrichment of any gene set by ORA for the recurrently affected genes, regardless of developmental cardiac expression gene filtering (all P > 0.05 before correction; Fig. 2, panels 3 and 4), suggesting stochastic effects of MDRVs in other genes in controls.
Fig. 2: Over-representation analysis (ORA) of recurrently affected genes identifies chromatin regulatory genes contributing risk to CTDs in 22q11.2DS.
Three different sources of gene sets totaling 19 are indicated by color below the bar graph (gene sets used by the PCGC to investigate sporadic CHD14 are indicated by black box, as well as in lilac and in gray). The first bar on the left in each panel shows the total number of recurrently affected genes (n) among all affected genes (N). The rest of the bars indicate the number of recurrently affected genes within each gene set (k) versus the final number of affected genes with MDRVs (most damaging rare variants) for each gene set (M) as indicated (see Methods for more details). The top two bar graphs show ORA results without filtering by gene expression levels in CTD cases (red) and with filtering (dark red) followed by the same for controls (green and dark green, respectively). The numbers in some of the bars denote the number of recurrently affected genes contained within the specific gene set / the total number of affected genes in this gene set. The gene set analyses were corrected for multiple testing by false discovery rare (FDR). Red asterisks denote significance after FDR correction; blue asterisk denotes borderline significance (P = 0.057).
Weighted gene set test expands chromatin regulatory genes identified in 22q11.2-CTD casesWe next employed a weighted gene set based test to evaluate whether high-confidence MDRV burden was significantly concentrated within any of the 19 gene sets. For this analysis, the mean expression level in cardiac progenitor cells from RNA-seq analysis of E10.5 mouse embryos was used. Again, we found chromatin regulatory genes contributing to 22q11.2DS-CTDs but identified a broader set of genes using this approach. The weighted gene set approach can reduce noise/signal ratio by prioritizing genes by their functional importance, and more specifically, by expression level during cardiac development, since genes in a gene set are not expected to contribute to disease equally. Genes affected with one or more MDRVs in one or more subjects, were included, thereby making it possible to expand the number of potential modifier genes. One gene set, Chromatin Regulatory Enzymes (274 genes), had significantly excess burden of MDRVs in 22q11.2DS-CTDs versus controls after false discovery rate (FDR) correction, with 24 identified MDRVs in cases and 14 MDRVs in controls (P = 5.00 × 10−4, Fig. 3, Supplementary Table 9); there was no significant enrichment of MDRVs in any of the three non-cardiac tissue gene sets (P > 0.05 before FDR correction). The MDRVs that each affect one or more chromatin gene is either ultra-rare (n = 12, AAF < 1.76 × 10−4) or novel (n = 30) in gnomAD (Supplementary Table 10). When taken together with ORA and weighted gene set approaches, we identified 42 MDRVs in 37 chromatin genes that occurred in 39, 22q11.2DS-CTD cases, accounting for 8.5% of 22q11.2DS-CTD cases in total. We found that 18 of the 37 chromatin genes are associated with known genetic syndromes that include CHD, supporting their candidacy (Supplementary Table 10).
Fig. 3: Identification of chromatin regulatory genes in 22q11.2DS by a weighted gene set approach.
Three different sources of gene sets totaling 19 are indicated by color (gene sets used by the PCGC to investigate sporadic CHD are indicated; black box). Y-axis in the left denotes the number represented by the bars (three bars per gene set): the total number of genes included in each gene set for the weighted analysis, the total number of 22q11.2DS-CTD cases and controls. Genes were weighted by gene expression level (blue dots indicate P-value; scale on Y-axis; red star is significant after FDR correction).
Overlap of chromatin regulatory genes between 22q11.2DS-CTDs with sporadic CHD, but differences in pathogenicity of variantsWe then compared the genes identified in 22q11.2DS-CTDs with chromatin regulatory genes found in cases of sporadic CHD from the PCGC. Sporadic CHD is defined by the PCGC as isolated cases in which neither parent is affected. Individuals with known syndromes, such as 22q11.2DS, were excluded, however subjects with isolated CHD or CHD that co-occurred with neurodevelopmental deficits or additional congenital anomalies were included21. Most of the chromatin regulatory genes found in studies by the PCGC had de novo mutations and most occurred in sporadic CHD with neurodevelopmental disorders or extracardiac features21. We cataloged genes that overlapped and found that 13 of the chromatin regulatory genes with MDRVs in the 22q11.2DS-CTD cases were identified to have de novo mutations (all categories: protein truncating, splicing, missense) in one or more subjects with sporadic CHD from the PCGC (total of 90 chromatin regulatory genes with MDRVs among 2871 CHD probands; Fig. 4a, b; Supplementary Table 11)7,21,22. In an independent WES study of sporadic CHD to identify genetic risk factors, Sifrim and colleagues examined sequence variants in similarly categorized individuals with sporadic CHD23. Nine genes in 22q11.2DS-CTD cases were found among 65 genes identified with de novo mutations in CHD with neurodevelopmental disorders or extracardiac features as shown in a Venn diagram (Fig. 4a) and UpSet plot (Fig. 4b)23,24 (610 subjects, syndromic-CHD; S-CHD cases; Fig. 4a, b). In an integrated analysis23, four chromatin genes in 22q11.2DS-CTD cases were identified among 16 found (16 de novo and inherited variants were found at the highest tier of significance among 1891 probands with/out neurodevelopmental disorders or extracardiac features in S-CHD vs nonsyndromic, NS-CHD; Fig. 4a, b). When taken together, 14 genes were shared among the 22q11.2DS-CTDs, PCGC and Sifrim et al. studies (CHD7, KAT6A, KMT2C, KMT2D, NSD1, SMAD4, TRRAP, EP400, IPO9, BRPF3, DNMT3A, HLTF, KDM4B, KMT2E, Supplementary Table 11), thereby providing further support of their role in cardiac development and disease. This finding is particularly compelling when considering that the identification of rare variants was performed using different patient cohorts (22q11.2DS-CTDs, sporadic CHD in general population by the PCGC7,21,22 and by Sifrim et al.23, study design (case-control, trios, and both trios and singletons, respectively), variant types (unknown inheritance for 22q11.2DS-CTDs, de novo and inherited recessive variants for sporadic CHD studies) and partially overlapping variant annotation pipelines/statistical methods. Further, of the 14 genes among the three studies, eight of ten that are causative of genetic syndromes have CHD as a notable feature25,26,27,28,29,30. Despite differences outlined above, overall, the focus of these studies was on rare variants associated with CHD that were deemed to be pathogenic, providing some similarities as well with the 22q11.2DS study (Supplementary Table 12).
Fig. 4: Chromatin regulatory genes shared with sporadic CHD in the general population.
a Venn plot showing the number of chromatin genes, as well as the number and P-value for the overlap (arrow) of chromatin genes identified between 22q11.2DS-CTDs (green) and studies of sporadic CHD (PCGC-lilac and Sifrim-dark blue is from integrated analysis, light blue is de novo mutations in S-CHD). A total of 1861 variants were found in 1261 genes serving as the background of the analysis. b UpSet plot illustrates the connections that are shown in the Venn plot. Sifrim CHD refers to Sifrim et al, CHD genes (n = 16); 22q11.2 chromatin refers to 22q11.2DS chromatin genes (n = 39); Sifrim chromatin refers to Sifrim et al, chromatin genes (n = 65); PCGC chromatin refers to PCGC chromatin genes (n = 90). Individual genes in each set are provided in Supplementary Table 11. c Types of variants in chromatin genes (PTV is protein truncating, D-mis are damaging missense variants, mis is missense). A total of 57 PTVs were identified in 90 chromatin genes in sporadic CHD by the PCGC. In total, 14 PTVs were identified among 24 de novo variants in nine genes by Sifrim et al. A total of three PTVs were found among 42 variants in 22q11.2DS-CTDs. P-values derive from two-Proportions Z-Test.
We next wanted to compare variants with pathogenicity identified in the clinical literature. Most of the MDRVs in 22q11.2DS-CTD cases were missense changes predicted to be damaging (Fig. 4c). From examination of clinical databases, 39 of 42 were considered variants of unknown significance (VUS; Table 2 and Supplementary Table 10). We then wanted to ask whether the types of variants in chromatin regulatory genes found in 22q11.2-CTDs might be similar to those found in studies of sporadic CHD. In contrast to 22q11.2-CTDs, protein truncating variants (PTVs; loss of function, LoF) were found more frequently in sporadic CHD by the PCGC (Fig. 4c)21,22,31. This was similar for studies by Sifrim et al.23 (Fig. 4c). Further, many of the variants identified in studies of sporadic CHD, in particular for chromatin regulatory genes, were de novo heterozygous mutations, but the variants we identified were of unknown inheritance. This suggests that variants in chromatin regulatory genes in 22q11.2DS-CTDs may affect gene function but are not disease causing on their own, thereby serving as modifiers.
Biological network connections between chromatin regulatory genes and TBX1To understand the biology of the genes, and with respect to TBX1, we generated a functional protein network using STRING software (https://string-db.org: Fig. 5a). The network consists of the 37 chromatin regulatory genes we identified plus two additional genes, CHD7 and ATAD2B, where we found MDRVs affecting more cases than controls in this study (two versus one, three versus two). As expected, there is an appreciable amount of functional coherence of these genes (P = 5.03 × 10−14). These chromatin regulatory genes have functions as histone lysine acetyltransferases and histone methyltransferases (Fig. 5b). We also identified sequence specific DNA binding proteins, suggesting cohesive but varied mechanisms by which these genes may increase risk for 22q11.2DS-CTDs (Fig. 5a, b).
Fig. 5: Chromatin gene network of modifiers for CTDs in 22q11.2DS.
a STRING image of 39 chromatin genes as identified in 22q11.2DS subjects with CTDs. Edges indicate both functional and physical protein interactions (Protein-protein enrichment p-value, 5.03 × 10−14). The types of interaction evidence for the network edges are indicated by the line color (text mining, experiments, database, co-expression, neighborhood, gene fusion and co-occurrence). The high confidence score of 0.700 was used to create the network. Kmeans clustering was used to generate three clusters (cluster 1, red; cluster 2, blue; cluster 3, green). The nodes based on confidence; with line thickness indicates the strength of the support of the data in STRING. Six genes found by ORA are indicated (Italic blue font for gene names), chromatin genes found with de novo mutations in one or more cases with sporadic CHD (with asterisk next to the gene names). Constrained genes are indicated (Black circle surrounding nodes) and candidate sporadic CHD genes are shown (underscore for the gene names). b Representative most significant gene ontology terms from the STRING image, color coordinated according to the STRING image (molecular function, MF; cellular component, CC; biological process, BP). FDR, false discovery rate, -log10 P-value.
A total of 22 of the chromatin regulatory genes and five of the six recurrent genes were among the most Constrained Genes gene set (Fig. 5a), and this overlap was significant (overlap P = 1.27 × 10−17 and 6.27 × 10−6, respectively). Additionally, nine are implicated in causing sporadic CHD from other studies in the literature21,22,32 as shown in Fig. 5a (overlap P = 7.05 × 10−5). Further, we show the overlap between the genes for sporadic CHD from the PCGC and the four from Sifrim et al. (Fig. 5a; Supplementary Table 11).
We next examined expression levels of the chromatin genes found in 22q11.2DS-CTDs in cardiac progenitor tissues. TBX1 is expressed in the progenitor cells of the pharyngeal apparatus (PA) that migrate to the cardiac OFT during early embryogenesis2. A logical hypothesis based upon results of this study is that altered chromatin regulatory genes in 22q11.2DS-CTDs may modify expression of TBX1 and/or downstream genes, disrupting normal cardiac development. If this is the case, such chromatin modifiers should be expressed in the same cell types as TBX1 during the same developmental period. Most chromatin regulatory enzymes are widely expressed but they may show enrichment in certain cell types or tissues. We therefore examined expression in the PA, OFT + RV and LV + atria. We found 36 of the 39 genes show enriched expression in TBX1 relevant tissues, specifically the PA as compared to either the OFT + RV or the LV + atria, where TBX1 is not expressed (Fig. 6; Supplementary Table 5) providing support for our hypothesis that chromatin regulatory genes might act in the genetic and epigenetic pathways of TBX1 function.
Fig. 6: Expression of chromatin regulatory genes is enhanced in cardiac progenitor cells of the pharyngeal apparatus.
Heatmap plot of RNA-seq results of genes from Fig. 5 by the log 2 transformed expression level in PA2-6, OFT + RV and LV+atria as indicated with highest red to lowest blue color. One gene, HNRNPD is significantly highly expressed than the rest of genes KPM = 507, 334 and 296, respectively, as the rest expression KPM value ranging from 1 to 197.
Comments (0)