This study was approved by the institutional review board (IRB) at the University of Texas Health Science Center in Houston (UTHealth) in accordance with the ethical standards of the Declaration of Helsinki. The research was conducted using the UK Biobank (UKB) Resource under Application Number 75470. The Penn Medicine BioBank (PMBB) is approved under the University of Pennsylvania IRB protocol #813913. The baseline demographic characteristics of this study’s cohorts are shown in Table 1.
Table 1 Baseline demographic data for cohorts included in studyUTHealth
DNA samples from affected individuals and relevant family members were collected after obtaining informed consent and human subject research approval. The UTHealth HTAD cohort includes families with two or more members affected by thoracic aortic disease, as well as trios of probands with aneurysm surgery or dissection at ≤ 40 years of age, with unaffected parents confirmed by imaging. The ESTAD cohort focuses on sporadic dissection cases in individuals ≤ 60 years of age without syndromic features or a family history. Genetic testing reports from patients with early onset sporadic aortic dissection undergoing clinical panel testing were obtained and reviewed. Exome sequencing (ES) was performed on the full HTAD and ESTAD cohorts, with select cases from unsolved HTAD pedigrees also undergoing whole genome sequencing (WGS).
UKB
The UKB data release from November 2023 (v18.1) was used for this study, including WGS data from approximately 500,000 participants20. Dissection cases were identified in individuals of European ancestry with an aortic dissection International Classification of Diseases, 10thRevision (ICD10) diagnosis or cause of death code (I71.0) or surgical code for aortic dissection (L27.4, L28.4) but without rupture (I71.1), resulting in 467 cases available for analysis. Similarly, individuals of European ancestry with a thoracic aortic aneurysm (TAA) were identified using the ICD10 code for thoracic aortic aneurysm, without rupture (I71.2). After excluding individuals with dissections as previously defined, a total of 1084 TAA cases remained for further analysis. A subset of 263 TAA patients requiring surgery was identified using surgical codes for open repair of the thoracic aorta or aortic root, excluding endovascular procedures. As a control cohort, the remaining 447,570 individuals of European ancestry without any ICD10 codes for aortic disease (I71) or congenital malformations, deformations and chromosomal abnormalities (Q00-Q99) were included for comparison.
PMBB
The PMBB is a genomic and precision medicine cohort comprising Penn Medicine health system patients who consent to linkage of electronic health records with biospecimens, including 43,731 who have undergone ES21. Thoracic aortic dissection was defined as having an encounter with an ICD10 diagnosis code of I71.01 or I71.03, or International Classification of Diseases, Ninth Revision (ICD9) codes 441.01 or 441.03. TAA was defined as having an encounter with an ICD10 diagnosis code of I71.1, I71.2, I71.5, or I71.6, or an ICD9 diagnosis code of 441.1, 441.2, 441.6, or 441.7 and excluding dissection codes.
SpliceAI Analysis
We limited our search to variants in HTAD genes that cause disease when only one functional copy is present (haploinsufficiency), namely FBN1 (NM_000138.5; MIM 134797), COL3A1 (NM_000090.4; MIM 120180), SMAD3 (NM_005902.4; MIM 603109), TGFB2 (NM_003238.6; MIM 190220), LOX (NM_002317.6; MIM 153455), and MYLK (NM_053025.4; MYLK 600922)3. For MYLK variants, only variants in the short isoform expressed in the thoracic aorta were considered22. In accordance with the Clinical Genome Resource (ClinGen) Sequence Variant Interpretation (SVI) Working Group guidelines10, we used a SpliceAI12 (version 1.3.1) score of ≥ 0.2 to identify non-canonical splice site variants that were likely to disrupt normal splicing. SpliceAI generates four scores - acceptor gain (AG), acceptor loss (AL), donor gain (DG), and donor loss (DL) - that indicate the likelihood of splice site alterations caused by a particular variant. For this study, when a given variant had multiple SpliceAI scores at or above the 0.2 threshold, we selected the maximum value for further analysis. We excluded homozygous variants and those with a minor allele frequency (MAF) ≥ 5 × 10−5 in the Genome Aggregation Database (gnomAD) version 4.1.023 as well as those with only Likely Benign or Benign ClinVar24 entries after 2019, the year SpliceAI was published. Variant pathogenicity was evaluated using ACMG/AMP and ClinGen guidelines10,11,25. Statistical differences between case and control groups were calculated with the two-tailed Fisher’s exact test using the Python SciPy library26, providing p-values, odds ratios (ORs), and 95% confidence intervals (CIs).
Sequencing
UTHealth ES was performed as previously described by Genome Sciences at the University of Washington27. Contemporaneously sequenced controls without aortic disease were provided. Briefly, exonic regions were captured using the SeqCap EZ Exome Library v2.0 from Roche. Enriched libraries were then sequenced on an Illumina NextSeq2000 using manufacturer protocols. Reads were mapped to the reference human genome (hg19) with BWA (Burrows-Wheeler Aligner)28, and variant detection and genotyping were performed using GATK Haplotype Caller29, with only variants passing all quality filters and having a minimum reported total depth (DP) of 20X being considered. After liftover to hg38 coordinates, variant call files (VCFs) were annotated with ANNOVAR30 for downstream analysis, including SpliceAI12 and gnomAD23 information. Additional WGS of select HTAD pedigrees was done at the University of Washington Center for Rare Disease Research as part of the NIH Genetics of Rare Disease (GREGoR) Consortium. After PCR-free library preparation of DNA samples, genome sequencing was performed on the Illumina NovaSeq platform, targeting an average coverage of 30X with 150 bp paired-end reads. After alignment to the hg38 reference sequence with BWA28, variants were called using the GATK HaplotypeCaller tool29, annotated on the SeattleSeq Annotation Server (http://gvs.gs.washington.edu/SeattleSeqAnnotation/), and analyzed on the open-source, web-based Seqr platform31. Sanger sequencing of UTHealth probands and any available affected family members was done to confirm variants identified through ES or WGS.
UKB WGS data processing was done using the UKB Research Analysis Platform (RAP) hosted on DNAnexus. Briefly, Illumina DRAGEN multi-sample pVCFs (hg38) were annotated with SnpEff and SnpSift32,33 for functional impact, gnomAD frequency, and SpliceAI information, and further analyzed with custom Python scripts.
PMBB ES data was processed as previously reported by Regeneron Genomics Center (RGC)34. Patient DNA samples were sequenced on the Illumina NovaSeq 6000 (Albany, NY, USA). WeCall variant caller was utilized for sequence alignment, variant identification, and assignment of genotype21. After quality control, single nucleotide variants (SNVs) were filtered for a read depth \(\ge\) 7 and were retained if they either had one or more heterozygous variant genotype with an allele balance ratio \(\ge\) 0.15, or a homozygous variant genotype.
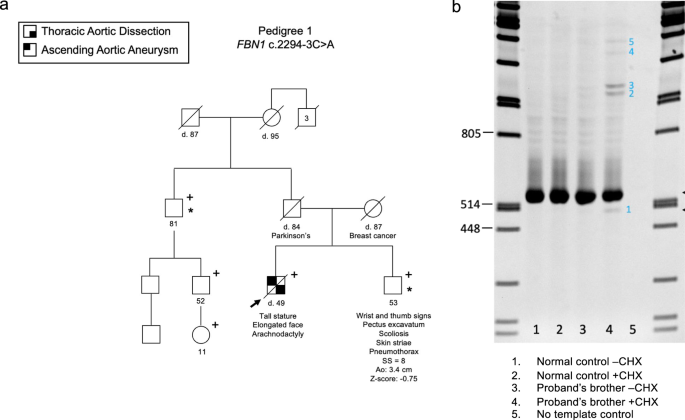
Splicing Assay (FBN1 c.2294-3 C > A)Cell culture and RNA Extraction
Dermal fibroblasts from a normal control and the individual with the FBN1 c.2294-3 C > A variant were grown, each in two 100 mm plates, to near confluence in DMEM with 10% FBS. One of two culture plates was incubated in the presence of cycloheximide (100 μg/ml, Sigma-Aldrich) for 6 hours before extraction of total RNA from both plates with the RNeasy Mini kit (Qiagen).
RT-PCR and Sanger Sequencing
Complementary DNA (cDNA) was synthesized with random hexamers and SuperScript™ III reverse transcriptase (Invitrogen). The FBN1 region of interest was amplified by PCR with a sense primer in exon 17 (5’- GAATGACGTCAGCAGGCAGT) and an antisense primer in exon 21 (5’- GGAGCAGCACTGGGACTTTA). The products were separated on 7% polyacrylamide gel, stained with ethidium bromide, and visualized using the “Carestream 212PRO” camera. The normal and all abnormal products were excised from the gel. The DNA was retrieved by submersion of the gel slices, separately, in 100 μl of sterile water at room temperature overnight, and 1 μl of each was reamplified using the same primers in exons 17 and 21. The amplicons were sequenced with BigDye™ Terminator v3.1 and capillary electrophoresis on the ABI 3500 Genetic Analyzer, and the data were analyzed with the Chromas software.
Splicing Assay (FBN1 c.7820-3 C > A)Cell Culture
Dermal fibroblasts from a patient with the FBN1 c.7820-3 C > A splicing variant and a gender and age-matched healthy control were grown in DMEM/High Glucose media (Hyclone) plus 10% FBS (Sigma) and antibiotic antimycotic solution (Sigma) in a 37°C, 5% CO2 incubator. After grown to confluence, patient and control cells were treated for 8 hours with either cycloheximide (100 µg/ml, Sigma) or DMSO (Sigma), as indicated.
RNA Extraction, RT-PCR and Sanger Sequencing
Total RNA from each plate was prepared with Trizol reagent (Thermo Fisher). cDNAs were generated using SuperScript IV VILO (Thermo Fisher). PCR products crossing the putative mutation site were amplified with primers E61-F (5’-CAGACCGGCTCCAGCTGTGAAGA-3’) and E65-R (5’-CATTGGCTTCTGTCTCAGACTG-3’) with KAPA HIFI PCR kit (Roche). The PCR products were Sanger sequenced with primers E61-Fa (5’-CCAGCTGTGAAGACGTGGAC-3’) and E64-Ra (5’-CAAGCCTCTGGGGAGAGTGA-3’).

Comments (0)