Study design
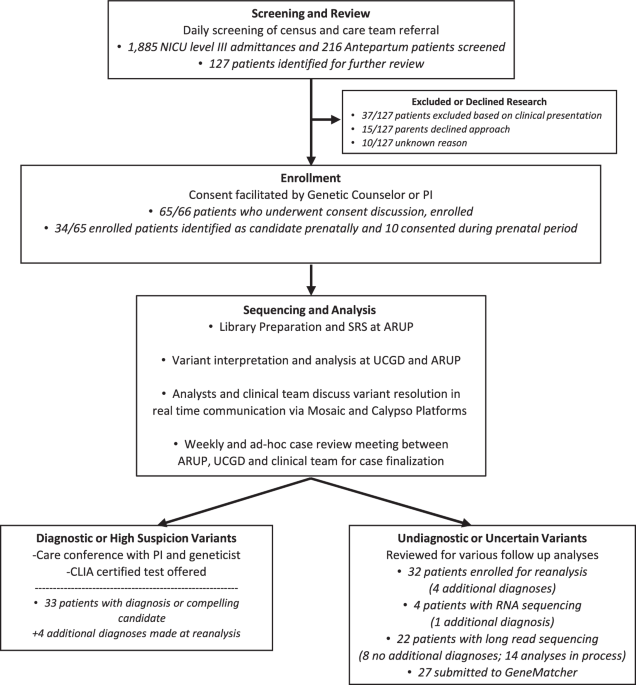
The purpose of the Utah NeoSeq Project is to provide diagnostic rapid whole genome sequencing (rWGS) to critically ill neonates and explore the utility and optimization of this technology. The study design of this project delivers a rapid return of results to clinical providers and families by operating under a research protocol, which allows for flexible and customizable research-based analysis workflows, comprehensive interrogation and reporting of all variant types, and prioritization of selected cases for experimental and analytical follow-up in the form of additional sequencing, clinical testing, or functional investigation of candidate variants. It was determined that a randomized control trial was not ethically appropriate given the growing evidence showing the improved outcomes and cost-efficiency of rWGS diagnosis in NICU populations. Therefore, all patients meeting the study criteria were offered enrollment. rWGS was completed in parallel with the clinical standard of care, which often includes sequencing of comprehensive phenotype-specific gene panels and/or chromosomal microarray analysis.
Ethical and legal considerations
We formed an expert panel of legal and ethical counsel, including members of the University of Utah Institutional Review Board (IRB), members of the University of Utah Center for Health Ethics, Arts and Humanities, pediatric legal counsel, genetic laboratory directors, and clinician researchers, with the goal of reviewing and evaluating applicable laws and weighing them with ethical considerations. Because rWGS was performed under a research protocol, we recognized that returning results prior to a Clinical Laboratory Improvement Amendments (CLIA)-certified confirmation could be interpreted as in conflict with CLIA regulations. Therefore, we created an IRB-approved protocol focused on informed consent in the setting of Health Insurance Portability and Accountability Act (HIPAA) right-to-access for families, to ensure that medically actionable variants with the potential for clinical intervention could be returned as research results22. Several factors were weighed in the development of this protocol, including the acute status of patients and the experience of our DNA extraction and research sequencing laboratory. Our consensus was that shared decision-making and transparency are fundamental to compassionate neonatal care. Parents were counseled regarding the limitations of genetic research results before and after testing, and the study team assisted in coordinating CLIA-certified confirmation testing for families with reported variants.
This research was conducted in full compliance with all relevant ethical regulations, including the Declaration of Helsinki. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Study investigators
Our study engaged three University of Utah-affiliated entities: ARUP Laboratories, a large non-profit University of Utah-affiliated reference laboratory, the Utah Center for Genetic Discovery (UCGD), an academic genome research center, and the University of Utah Department of Pediatrics. This collaboration included laboratory personnel and directors, genome scientists and analysts, molecular geneticists, and clinician researchers from Neonatology, Medical Genetics, Maternal Fetal Medicine, and other pediatric sub-specialties. Collaborations were established with the University of Utah Model Functional Analysis Service and the Utah Clinical and Translational Science Institute (CTSI), allowing for ongoing investigation of candidate variants.
Population, recruitment, and consent
Acutely ill infants suspected by their treating clinicians of having a genetic etiology were offered enrollment, along with their biological parents. Common indications included multiple congenital anomalies, unexplained seizures, arthrogryposis/skeletal dysplasia, non-immune hydrops fetalis, and acute clinical presentation and hospital course out of proportion to expectations in the NICU. Neonates with an established non-genetic cause for presentation, a known genetic diagnosis, or with high suspicion for aneuploidy were excluded from the study. We identified and enrolled patients both prenatally and postnatally. Prenatally, we identified patients and consented through the fetal care center and through the University of Utah hospital, which specializes in high-risk deliveries of patients who will require Level IV care at Primary Children’s Hospital. Postnatally, we evaluated infants at our inborn Level III NICU. Clinical providers aided in the identification of potential patients and introduced the idea of genetic testing to the families.
Our consent22 (Supplementary Note 1) was offered in both English and Spanish, with short forms available for 61 additional languages. We required an explicit opt-in or opt-out to receive incidental “medically actionable” findings for the patient and each parent. The consent outlined that participants’ samples and information would be stored indefinitely for further research. If new results are identified through future research, participants are contacted and asked if they would like to receive these findings, with the understanding that their interest in research participation may have changed since they were initially consented.
Sample collection and sequencing
Proband umbilical or venous blood and parent venous blood were collected and couriered to ARUP Laboratories, typically within 1 hour. Genomic DNA was extracted from 0.5 to 1 mL whole blood using the Chemagic Magnetic Separation Module I (PerkinElmer, MA, USA) and quantified with a broad-range double-stranded assay kit on a Qubit 1.0 fluorometer (ThermoFisher Scientific, USA). For forty-three families, sequencing libraries were prepared with the Illumina DNA Prep workflow (Illumina, CA, USA). Dual-indexed, paired-end, whole genome libraries were prepared from 500 ng input DNA using on-bead transposons to normalize the DNA fragmentation and adapter ligation process. The normalized product underwent five PCR cycles to add unique 10-bp dual indices and sequencing adapters. After amplification, double-sided bead selection was used to select appropriately sized library fragments. The double-stranded libraries were quantified via electrophoresis on the 4200 TapeStation with high-sensitivity D5000 tapes (Agilent, USA), diluted to 3 nM, and pooled at equimolar ratios. The final library pool was diluted to 1.6 nM and spiked with 1% PhiX bacteriophage DNA as a sequencing control (Illumina, CA, USA)11. For twenty-two families, libraries were prepared for sequencing with the Illumina PCR-Free Prep workflow (Illumina, CA, USA) from 1 µg input DNA. Adapters, including 10-bp dual indices, were added via ligation. The final single-stranded libraries were pooled at fixed volumes and quantified via single-strand DNA Qubit (ThermoFisher, USA). These final pools were diluted to 2.6 nM and spiked with 1% PhiX (Illumina, CA, USA). For both library preparations, the final pool was denatured and loaded on a NovaSeq 6000 instrument for paired-end sequencing (2 × 150 bp) on an S1 flow cell (Illumina, CA, USA). Sequence reads were demultiplexed and converted to FASTQ files with bcl2fastq (v2.20) and securely transferred from ARUP to UCGD.
BioinformaticsSNV & indel variant calling
Automated calling of single nucleotide variants (SNVs) and short insertions and deletions (indels) was performed by the UCGD Core. The SNV/indel calling pipeline used the FastQForward parallelization platform. FastQForward wraps the BWA short-read aligner23 and the Sentieon24 variant calling toolkit, a computationally efficient re-implementation of GATK algorithms, for an overall workflow that follows GATK best practices25. Reads were aligned to a modified version of the GRCh38 reference genome that has false segmental duplications and other known erroneous sequences masked out by Ns, as per recommendations issued by the Genome Reference Consortium and Telomere-to-Telomere (T2T) Consortium. Variants were annotated using VEP, including five splice prediction plug-ins (SpliceAI, MaxEntScan, SpliceRegion, dbscSNV, and GeneSplicer), and the UTRannotator plug-in Automation of the NeoSeq pipeline used NICUWatch (https://github.com/srynobio/NICUWatch), a codebase to manage sample data transfer, multi-platform project creation, workflow implementation in Nextflow26, and Amazon SNS messaging service to inform team members of data processing status and enable rapid downstream analysis.
Structural variation
Structural variants (SVs) were identified using Smoove27 (https://github.com/brentp/smoove), Manta28, and RUFUS (https://github.com/jandrewrfarrell/RUFUS). Smoove and Manta call structural events using split-mapped reads and discordant read pair insert sizes. SV calls were annotated with overlapping genes and duphold metrics29 using Smoove, and with population allele frequencies using SVAFotate30. On select cases, data were re-aligned to the gapless T2T reference genome and reanalyzed with Smoove for further investigation of ambiguous SV calls. RUFUS performs de novo assembly of reads containing sequence kmers unique to the proband and absent from one or more parental controls, creating assembled contigs of mutant haplotypes and allowing identification of complex structural rearrangements as well as SNVs and indels. Mixed-type compound heterozygotes consisting of an SNV/indel and SV in trans were identified using RUFUS as well as manual comparison of SNV and SV candidate lists.
Other variant types
Short tandem repeat expansions at known disease-associated repeat loci were identified using GangSTR31 and STRling32 and filtered using dumpSTR and custom scripts. Aneuploidies and large copy number variants were identified using MoChA33 and a RUFUS derivative that examines genome-wide kmer depth (unpublished). Uniparental disomy was identified by examining per-chromosome Mendelian inheritance error rates using MoChA. Extended regions of homozygosity indicative of monosomy, large deletions, consanguinity, or uniparental isodisomy were identified using the ‘bcftools roh’ subcommand34.
Variant selection and prioritization
We used Slivar35 to filter for high-impact, rare variants consistent with the possible modes of inheritance suggested by the pedigree structure. High-impact variants included nonsynonymous variants, splice region variants, intronic and synonymous variants predicted by any of five splice prediction programs to alter splicing, and 5’ UTR start-gain variants. We also used the statistical approaches of VAAST36,37 and Phevor38 via Fabric Genomics and GEM39 to prioritize variants according to predicted deleteriousness, conservation, population allele frequency, known associations of the gene to Human Phenotype Ontology (HPO) terms assigned to the patient, and other prior information about the specific variant (e.g. ClinVar classifications).
Quality control
Quality metrics were generated at various stages of the analysis. FASTQ files were evaluated for read quality using the fastp40 tool. Alignments were evaluated for per-chromosome coverage using ‘goleft indexcov’41 and for general alignment metrics using alignstats (https://github.com/jfarek/alignstats). Variant calls were evaluated using ‘bcftools stats’42 for variant statistics and Peddy43 to identify issues with assigned relatedness, sex, ancestry, and DNA contamination. Select QC results were aggregated and visually displayed for review using MultiQC.
Visual data review, communication, and project management
The software tool Mosaic (https://frameshift.io/) from Frameshift Genomics was used to support multiple aspects of project management, phenotype presentation, analysis, data quality and results review, and collaboration for team members. Extended candidate variant lists, prioritized gene lists, and detailed quality assessment data were uploaded and displayed within the tool, and its visualization functionality was used to compare data across NeoSeq cases. Mosaic also provided secure access to the underlying genomic data, allowing tools such as IGV and iobio13,44,45,46,47 to be dynamically launched and saved.
Reporting and confirmation
The multidisciplinary team, consisting of clinicians, laboratory medical directors, and analysts, met weekly to review cases. Candidate variants were classified using the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) guidelines used for CLIA-certified clinical testing, and all reported variants were reviewed and signed out by an American Board of Medical Genetics and Genomics (ABMGG)-certified clinical molecular geneticist, a medical geneticist, and the study PI48. Variants were reported in the categories of diagnosis, selected VUS, and medically actionable incidental findings (for those patients who opted to receive such information during enrollment). VUS were reported when there was significant phenotypic overlap, a high suspicion of pathogenicity, or when pathogenicity could potentially be resolved through additional family testing or non-invasive patient workup. For diagnostic findings, the clinicians informed interpretation based on clinical presentation; sub-specialist reviewers were available from all pediatric divisions servicing the NICU in order to allow for precise clinical correlation. Medically actionable incidental findings included but were not limited to all ACMG-recommended secondary genes; it was determined that the ‘medically actionable’ status of variants should be evaluated on a case-by-case basis with consideration of current gene-disease associations and developments in management and therapies. Pathogenic and likely pathogenic variants were reported for incidental findings in line with ACMG guidelines49.
Two documents were generated by the study team for each case: a research report and a results letter. The research report was a technical summary of all candidate variants that were identified and discussed, including variants that did meet the criteria for return to families and was kept as an internal reference for the analysis team to document evidence for and against pathogenicity and to consult upon yearly re-analysis. The results letter contained returnable results, was written in language intended for providers and patients, and was uploaded to the patient’s electronic medical record under the research section and labeled as research results. The study team determined if additional follow-up was needed, including family genetic testing or biochemical laboratory testing, and these recommendations were included as a section in the result letter. Results were shared with the clinical care providers and returned to the parents by the study neonatologist and medical geneticist. Reported variants were clinically validated with CLIA-certified genetic testing, when available, and were confirmed using non-WGS research methods when CLIA-certified testing was not available.
Periodic reanalysis
Cases without a diagnostic result underwent yearly data re-analysis with a review of recent literature and medical records to refine the phenotype. Periodic re-analysis is considered imperative in the neonatal population, given that many phenotypes that inform analysis may not appear until later in childhood. For candidate variants in genes of uncertain significance to human disease, we utilized GeneMatcher, a freely accessible web service that allows individuals to post a gene of interest and connects them internationally with other individuals who post the same gene, facilitating comparisons of variant type and clinical presentation if available.
Long-read sequencing
Diagnostically negative cases were candidates for long-read WGS. Long-read libraries were prepared from 5 µg genomic DNA using the standard PacBio HiFi protocol (PacBio, CA, USA). Genomic DNA was sheared on a MegaRuptor 3 (Diagenode) to 15–20 kb fragments. Prepared libraries were size-selected on a 0.75% BluePippin gel (Sage Sciences). Libraries were quantified using a high-sensitivity double-stranded DNA kit (ThermoFisher, USA) and size checked with a Genomic DNA 165 kb Kit on the Femto Pulse (Agilent, USA). HiFi Sequencing was performed on the Sequel IIe (PacBio, USA). Probands were sequenced with three 8 M SMRT cells, and parents were each sequenced with one SMRT cell. All SMRT cells had 30-hour movies collected. HiFi reads were aligned to the modified GRCh38 reference using pbmm2, and SNV/indels were identified using DeepVariant50 and joint called using Glnexus. Short-aligned Illumina reads were then merged with aligned HiFi reads, and SNV/indels were recalled using DeepVariant’s HYBRID_PACBIO_ILLUMINA model. Variants were annotated using the same protocol as with short reads. Structural variants were identified with pbsv using only HiFi reads.
RNA Sequencing
For selected diagnostically negative cases and cases with a candidate variant predicted to affect splicing, participant blood was drawn into a PAXgene Blood RNA Tube (IVD) (Qiagen: 762165). Cellular RNA from stabilized blood was isolated and purified using PAXgene Blood RNA Kit (Qiagen: 762164), including DNase I treatment to remove any residual DNA. Libraries were prepared using the Illumina TruSeq Stranded Total RNA Library Prep with Ribo-Zero Globin kit (20020613) with TruSeq RNA UD Indexes (20022371). Sequencing libraries were chemically denatured and applied to an Illumina NovaSeq flow cell at 1.3 nM using the NovaSeq XP workflow (20043131), and a 150 × 150 cycle paired-end sequence run was performed on a NovaSeq 6000 instrument using a S4 reagent Kit v1.5 (20028312).
RNA analysis
RNA sequence reads were aligned with STAR51 using twopassMode=Basic to produce aligned BAM files and junction files. LeafCutterMD52 was run on the indexed BAM files for all patients and parents currently sequenced (48 samples in total) to identify candidate outlier splice junctions for each patient. A custom python script was used to modify the STAR-generated junction files into a form compatible with IGV53 and IOBIO13,44,45,46,47. Deeptools bamCoverage54 was used to generate bigwig files representing read coverage in aligned BAM files. Custom Perl scripts were used to split LeafCutterMD results by patient and format them as BED files. Bedtools55 was used to annotate the candidate splice junctions from LeafCutterMD with overlapping gene symbols. We collected candidate genes that had the potential to be affected by altered splicing from both identification of splice site/region variants from our WGS analyses and splice outlier candidates from the LeafCutterMD analysis. These RNASeq candidate genes were evaluated for overlap between the phenotypes described in the HPO for each candidate gene and the phenotypes derived from natural language processing-based extraction of HPO terms from the patient’s medical records using ClinPhen56. Phenotype overlap was scored by a custom python script that implements a semantic similarity search similar to that used by Phenomizer57 and originally described by Resnik et al.58. Candidate splicing events that impacted genes with phenotype overlap scored in the top 10th percentile for all genes annotated by HPO with disease associations were manually reviewed. Manual review of final candidates consisted of evaluating the read data supporting putative splicing events by viewing the RNASeq alignments supporting the event in IGV and the viewing the RNASeq junction files and bigwig coverage files in a custom IOBIO13,44,45,46,47 application designed for reviewing splicing events. Genes and associated splicing events that were well supported by both phenotypic overlap and RNASeq read support were reviewed for clinical relevance by the full NeoSeq team. The steps in the analysis pipeline above were managed with a Snakemake59 workflow.
Functional analysis evaluation
As needed, NeoSeq cases were reviewed by a functional analysis team consisting of collaborating investigators with expertise in functional genetics in cell lines, Drosophila, and zebrafish, in assessing the consequence of candidate variants in various model systems. Clinical and genetic information was integrated with a database and literature review to determine the most informative and feasible opportunities for experimental exploration of potential diagnostic variants.
Measuring outcomes and utility of testing
Parental satisfaction and understanding are crucial for enhancing the consent process for future parents and may increase the inclusion of more diverse participants in whole genome sequencing studies. We surveyed parents (Supplementary Note 2 and 3) who either participated in or declined participation in the study to evaluate their attitudes toward consent, research participation, satisfaction with genomic testing, and experiences of decisional conflict and regret. Following the consent process, or the decision to decline, the study coordinator approached parents to invite them to participate in a voluntary survey. This survey was conducted at the child’s bedside and took ~3–5 mins to complete with the assistance of the study coordinator. Participants were able to skip any questions they did not feel comfortable answering. Surveys used multiple-choice, open-ended answers, and the Likert scale.
We also conducted a survey (Supplementary Note 4) among clinical providers to assess the utility of rWGS in the diagnosis, management, and counseling of their patients. The survey was distributed via REDCap within 5–7 days following the return of NeoSeq results. A cover letter accompanying the survey outlined its purpose, and consent was implied through the completion of the survey. The survey, estimated to take ~5 minutes, included questions regarding changes in clinical management, the perceived utility of rWGS testing, and its perceived impact on the family.

Comments (0)