
Remember me
Causative mutations in MECP2 have been identified in 90–95% of classic RTT cases and 50–70% of atypical RTT cases6,7. However, many clinically evident RTT or RTT-like cases remain with no molecular diagnosis. Identifying the genetic basis of RTT cases is crucial, as it provides families with a definitive diagnosis of their child’s condition, alleviates uncertainty and anxiety regarding future pregnancies, and allows for appropriate medical management tailored to the underlying genetic cause – including opening opportunities to participate in clinical trials targeting MECP2-specific pathways. Many other clinical conditions can imitate cases of Rett syndrome18,19, and misdiagnosis may lead to ineffective or potentially harmful treatments. Thus, uncovering the genetic underpinnings of these cases is essential for optimizing patient care and advancing prevention and treatment strategies.
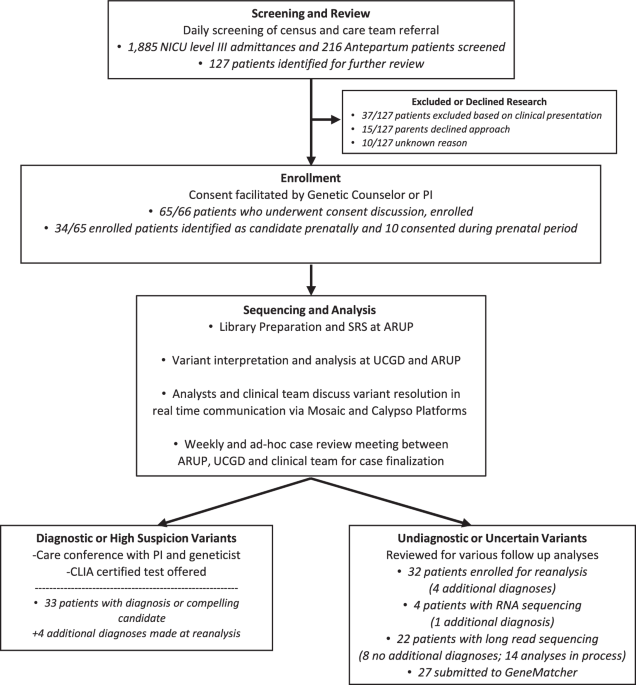
At the Israeli Rett clinic at Sheba Medical Center, 225 patients with a clinical diagnosis consistent with Rett syndrome are being followed20,21. Among these, 10 patients have remained without a molecular diagnosis for years. We set out to solve the genetic riddle of 3 of these cases: two with typical RTT and one with the atypical preserved speech variant (PSV). These patients were meticulously clinically diagnosed by a highly experienced pediatric neurologist specializing in Rett syndrome, but had no genetic diagnosis despite extensive testing over many years. We solved all three cases using WGS, revealing that each was caused by a distinct SV within MECP2.
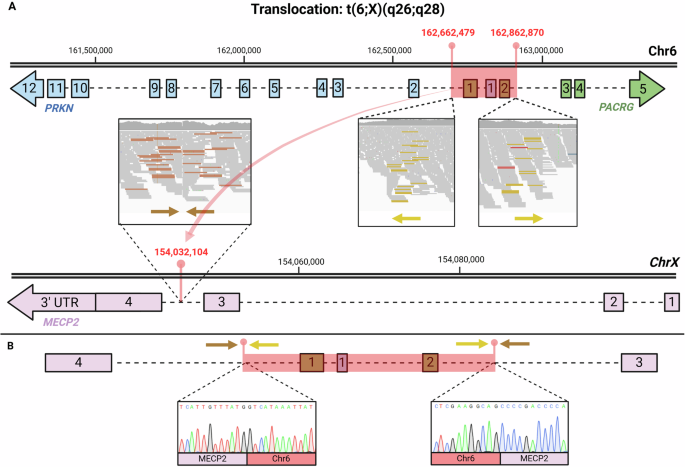
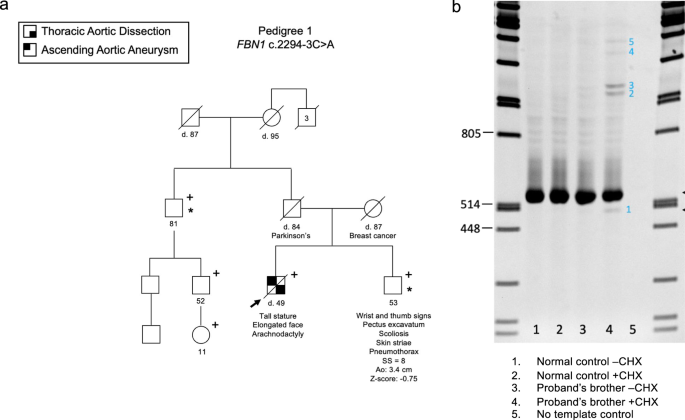
We demonstrated that all three RTT cases were caused by disrupting both of the main isoforms of MECP2: MECP2_e1 (NM_004992.4) and MECP2_e2 (NM_001110792.3)22. In Case 1, we identified a ~ 200 Kbp translocation from chromosome 6 to the X chromosome; t(6;X)(q26;q28). The translocation breakpoint within MECP2 (hg38: chrX:154,032,104) is located between exons 3 and 4 (Fig. 1). This ~200 kbp insertion encompasses the complete exons 1 and 2 of PACRG and the entire exon 1 of PRKN (Fig. 1). We hypothesize that this translocation may disrupt normal splicing, potentially triggering nonsense-mediated decay (NMD) or leading to the production of an aberrant MECP2 protein. We consider it highly unlikely that this insertion within the core region of MECP2 would result in the synthesis of a normal WT protein. In Case 2, we found a cSV involving a large deletion that removes the entire downstream portion of both MECP2 isoforms, including exons 3 and 4, as well as the entire 3’ UTR (Fig. 2). This likely results in the transcript undergoing NMD, preventing protein production. In Case 3, we identified an approximately 3,200 bp deletion in the downstream region of MECP2 (NC_000023.11:g.154027486_154030665del). This deletion removes the terminal portion of exon 4, resulting in a stop-loss mutation and it also includes a 2,889 bp of the 3’ UTR (Fig. 3). The deletion affects amino acids 397 to 498 and causes a frameshift, generating a novel amino acid sequence of 149 residues before encountering a premature stop codon.
SVs are genetic structural alterations, typically defined as genomic variants larger than 50 bps. These variations can include deletions, duplications, insertions, inversions, and translocations, which may impact gene function and regulation. SVs can significantly impact health by disrupting gene function, leading to a wide range of diseases, including developmental disorders, intellectual disabilities, and various congenital conditions23,24,25. Small-intermediate SVs typically range from 50 bp to 50kbp in size, whereas large SVs encompass alterations exceeding 50kbp26,27.
Small-intermediate SVs pose a unique challenge in detection compared to single nucleotide variants (SNVs), indels, and large SVs, as they fall within a range where conventional sequencing methods may fail to accurately identify and characterize due to limitations in read length and resolution methods23,28,29. Short read sequencing (SRS), in particular, encounters significant difficulties in detecting small-intermediate SVs29,30. This is because it often fails to span the exact SV breakpoints and map them accurately, hindering the detection of such variants, especially those located in non-coding regions or characterized by combinations of structural changes, known as cSVs30. Additionally, relying solely on SV detection software is complex, as it typically identifies thousands of SVs per genome, many of which are false positives or inaccurately predicted variants31,32.
Since the discovery of Rett syndrome by Andreas Rett in 1966, numerous cases caused by MECP2 mutations have been solved using conventional methods. SNVs, indels, and copy number variants (CNVs) were mostly detected via routine genetic panels, whole exome sequencing (WES), and directed targeted sequencing of MECP233,34,35,36,37,38,39. Large SVs were primarily detected using chromosomal microarray (CMA) or fluorescence in situ hybridization (FISH)40,41,42,43,44,45,46. Multiplex ligation-dependent probe amplification (MLPA) identifies large deletions35,47, that might be missed by routine PCR-based screening strategies. For example, one group succeeded in detecting deletions ranging from 1235 bp to 85 kb within MECP2 using MLPA48; but, to characterize the rearrangements and locate the exact nucleotide positions of the breakpoints, they had to use real-time quantitative PCR (qPCR) and long-range PCR. Therefore, it is understandable that general screening of the Human Gene Mutation Database (HGMD)49 has shown these small to intermediate SVs to be much less recognized over the years compared to other types of mutations.
We present an effective approach that allowed us to uncover novel disease-causing SVs in MECP2 using conventional short-read WGS. In two of the three cases (Cases 1,2), MANTA software facilitated the identification of potential SV breakpoints within MECP2. MANTA software can detect discordant read pairs, split reads, and abnormal read depth, which are indicative of various SVs such as deletions, duplications, inversions, and translocations. By integrating these different types of evidence, MANTA can suggest genomic loci suspected to be SVs breakpoint borders. However, due to MANTA’s tendency to produce false positives and its limitations in predicting exact SV subtypes, especially cSVs, we found it necessary to directly analyze and visualize the BAM file alignment in the regions that MANTA recognized. We utilized the IGV to visualize read mapping, which enabled us to interpret and hypothesize what were the distinct SV types in each case, and accurately determine their respective boundaries. The utility of IGV was highlighted when Case 3 was resolved solely through visualizing MECP2 on IGV, bypassing the need for MANTA. Eventually, the borders of identified SVs that we hypothesized to exist were verified by PCR and Sanger sequencing. That approach enabled us to detect pathogenic novel, elusive small to intermediate SVs within MECP2, that evaded detection using any other diagnostic tool (such as CMA, Trio exome, RTT-like genes panel, MLPA, etc.).
It is worth noting that cSVs pose great challenges in SRS mapping interpretation, beyond those in simple SVs. The presence of multiple SVs within the same genomic region can obscure and impede their identification. For instance, in case 2, the deletion sites were not apparent in the BAM visualization due to the presence of the inversion between them. We could infer the presence of deletions only by speculating on what would be logical and how the paired-reads might align, assuming there might be an inversion. Our findings highlight the importance of direct searching for SVs when conventional methods prove insufficient in detecting any mutations in MECP2 in RTT cases48,49.
Long-read sequencing (LRS) is effective in identifying disease-associated SVs and cSVs50. However, its high cost makes it impractical for routine genetic testing. As we demonstrate, such cases can be effectively resolved through advanced cost-effective bioinformatics analysis tools designed to detect SVs in SRS data. Our findings are consistent with a previous report that identified a 2.6 kb intronic insertion variant within MECP2 using MANTA software51. Additionally, our results demonstrate that even manually scanning IGV with a targeted focus on a specific gene can lead to the identification of SVs. We have shown that simply visualizing and carefully examining the MECP2 gene in the patient’s BAM file using IGV software can reveal inappropriately mapped reads that may indicate the presence of a disease-causing SV. Despite the obvious need for such software, there are currently no user-friendly and reliable bioinformatics tools available for routine use in identifying SVs in MECP2, nor is there a standard practice for directly examining the MECP2 gene in BAM files from Rett patients. This gap highlights the need for developing cost-effective and accessible techniques to improve the diagnostic process for SV detection.
The clinical phenotypes associated with MECP2 mutations exhibit significant variability, and prior studies have explored whether this variability is influenced by the type and location of the mutation52,53,54. Bebbington et al. developed a phenotypic profile of C-terminal deletions in Rett syndrome, finding that such cases often present with milder disease phenotypes55. These individuals are more likely to have normal head circumference and weight, a later onset of stereotypies, and earlier acquisition of walking skills. Additionally, deletions occurring downstream within the MECP2 gene were associated with lower average severity scores compared to those occurring upstream55. However, the phenotypes observed in our cases diverge from this typical profile; in Case 3, we identified a deletion at the C-terminus of MECP2 (Fig. 3), resulting in a frameshift that alters the protein sequence starting at amino acid 397 and introduces a stop codon at position 545. Despite this C-terminal mutation, the patient’s phenotype aligns with classic Rett syndrome, except for the absence of seizures. She presented with microcephaly and has not achieved independent walking, which contrasts with the typically milder phenotype associated with C-terminal mutations. Similarly, in Case 2, the mutation involved a cSV, with a larger deletion encompassing exons 3 and 4 and the entire 3’ UTR (Fig. 2), and the patient also exhibited features consistent with classic Rett syndrome. Interestingly, in Case 1, the patient was diagnosed as PSV, a milder phenotype previously described in the literature56. Given the large translocation identified in this case, a classic Rett syndrome phenotype might have been expected. While there are occasional genotype-phenotype correlations linked to the position of the mutation, other factors—such as the X-inactivation ratio, modifier genes, and additional, less well-known epigenetic influences—also contribute to phenotypic differences and severity57,58,59.
Case 1 also raises the question of whether the two disrupted genes on chromosome 6 contribute to the patient’s phenotype. As described, the balanced translocation breakpoints in this case are located within two genes on chromosome 6: PRKN and PACRG. PRKN is primarily associated with Parkinson’s disease in the biallelic state; in this case, however, it is disrupted in a heterozygous state, which is not typically linked to the disease. Similarly, according to OMIM60, PACRG has not been associated with any disease in the heterozygous state. The translocation site within MECP2, however, lies in a critical region that likely impacts the MECP2 transcript relevant to Rett syndrome (NM_004992.4, MECP2_e1), suggesting it is a primary contributor to the patient’s phenotype.
To date, dozens of variants in several genes have been suggested to be causative of Rett and Rett-like syndrome in cases where no MECP2 mutation was found13,14,61,62. It is plausible that some of those proposed variants are not disease-causing, as causative small or intermediate SVs within MECP2 have possibly eluded detection, or because there may be a double diagnosis involving both MECP2 SVs changes and a pathogenic mutation in another gene, leading to a more complex phenotype.
We propose the following approach to address elusive SVs and cSVs cases (Fig. 4): in clinically diagnosed RTT cases without a detectable MECP2 mutation, it is crucial to investigate the presence of SVs. LRS or WGS should be considered as diagnostic tools. If feasible, LRS is preferred. When using WGS, SV breakpoints should be sought within the MECP2 gene. The patient’s BAM file can either be manually scanned or analyzed with SV detection tools such as MANTA, Delly, and others. Visualizing read mapping by IGV may uncover subtle clues within the SRS data that aid in accurately identifying SV boundaries. Finally, these identified SV breakpoints can be validated through PCR and Sanger sequencing.
Fig. 4: Approach for uncovering elusive novel disease-causing structural variants (SVs) in genetically unsolved Rett syndrome (RTT) cases.
LRS; Long-read sequencing. SVs; Structural variants. IGV; Integrative Genomics Viewer.
This methodology enabled us to successfully resolve all three investigated RTT cases. We believe that this approach could be valuable for resolving SV cases in other diseases, as these types of variants are not exclusive to Rett syndrome. We suggest that this approach should be considered for any genetic case exhibiting a distinctive phenotype with a limited number of related genes, or where there is a confined linkage area. In such instances, it would be feasible to avoid thousands of false positive variants and to investigate small to intermediate SVs within these specific loci or genes.
As we investigated 3 cases with a clear RTT phenotype, unraveling causative SVs in all 3 cases, it is plausible that SVs possibly constitute a common cause of RTT in yet unresolved cases. We have succeeded in elucidating the pathogenic SVs through tedious work using existing software and IGV visualization and interpretation. However, this process can be greatly facilitated through the generation of user-friendly clinical analysis software that would enable clinicians of various disciplines, not necessarily geneticists, to easily filter VCF files in search of SVs. We propose that it may be feasible to develop such software by integrating different programs capable of identifying suspected SV regions and combining them with the ability to filter by various loci, genes, or related phenotypes using databases such as OMIM or HPO. Such software could be routinely used and facilitate the resolution of many elusive SV and cSV cases, not only in Rett syndrome but also in other monogenic diseases.
Comments (0)