
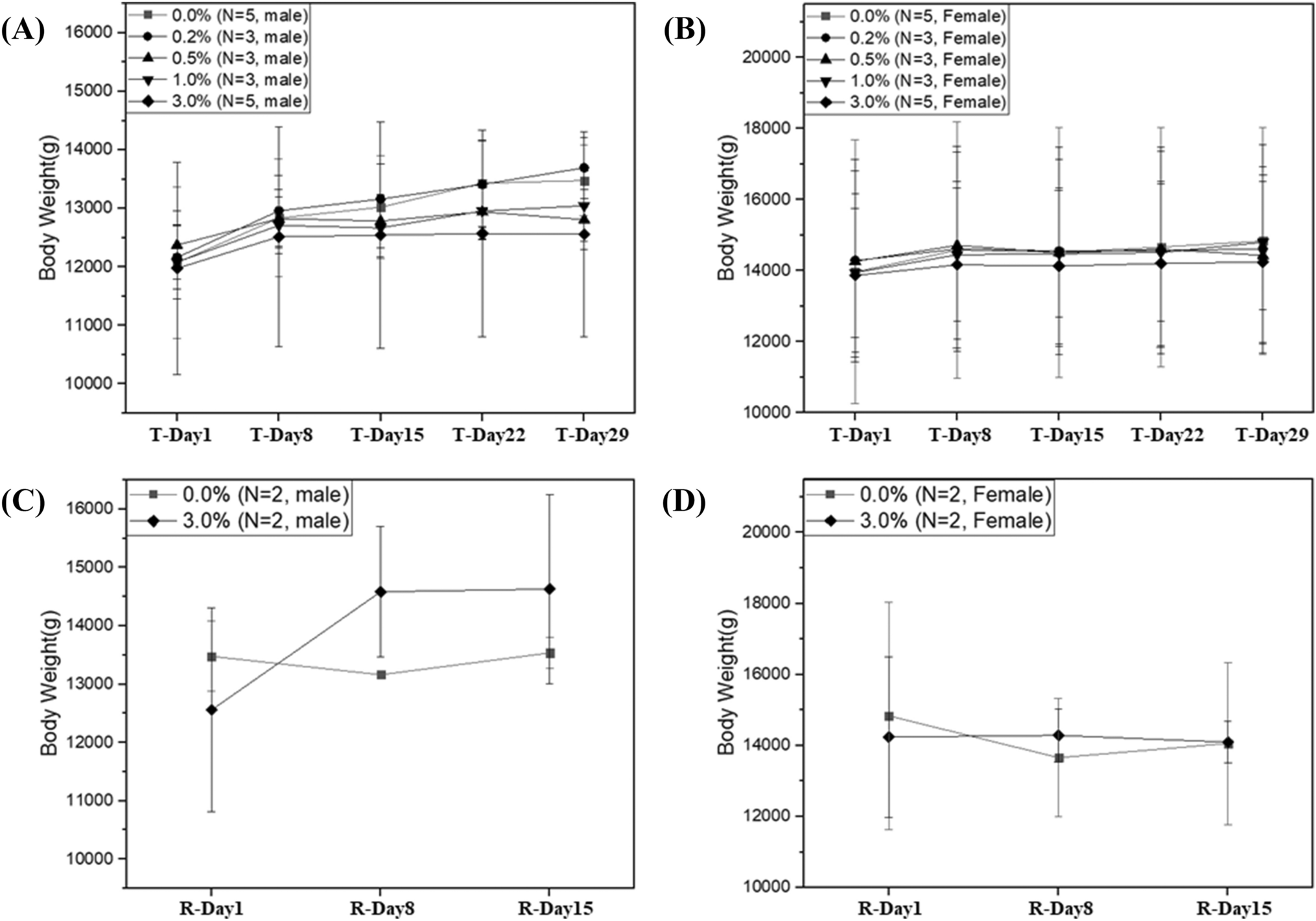
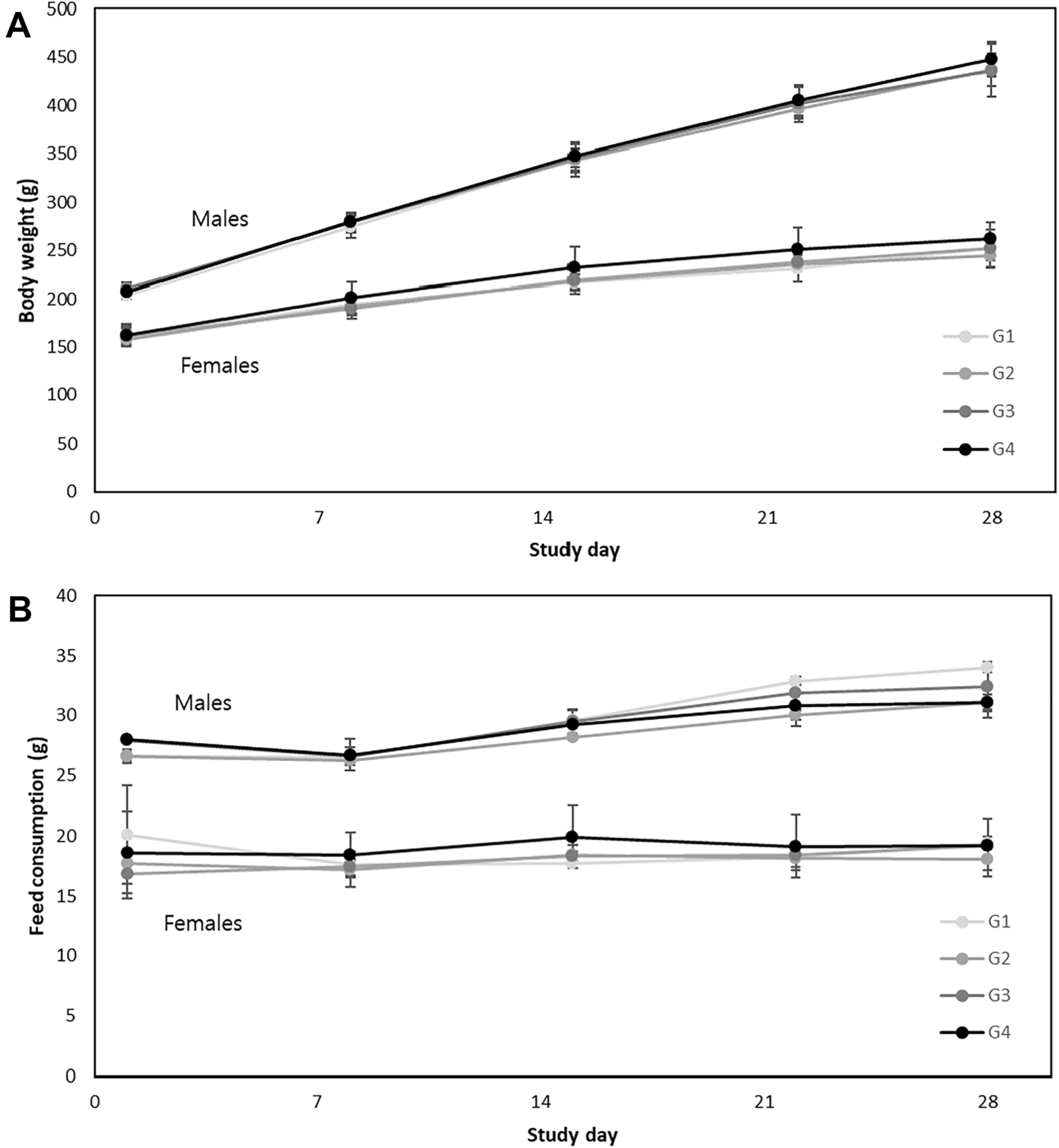
Remember me
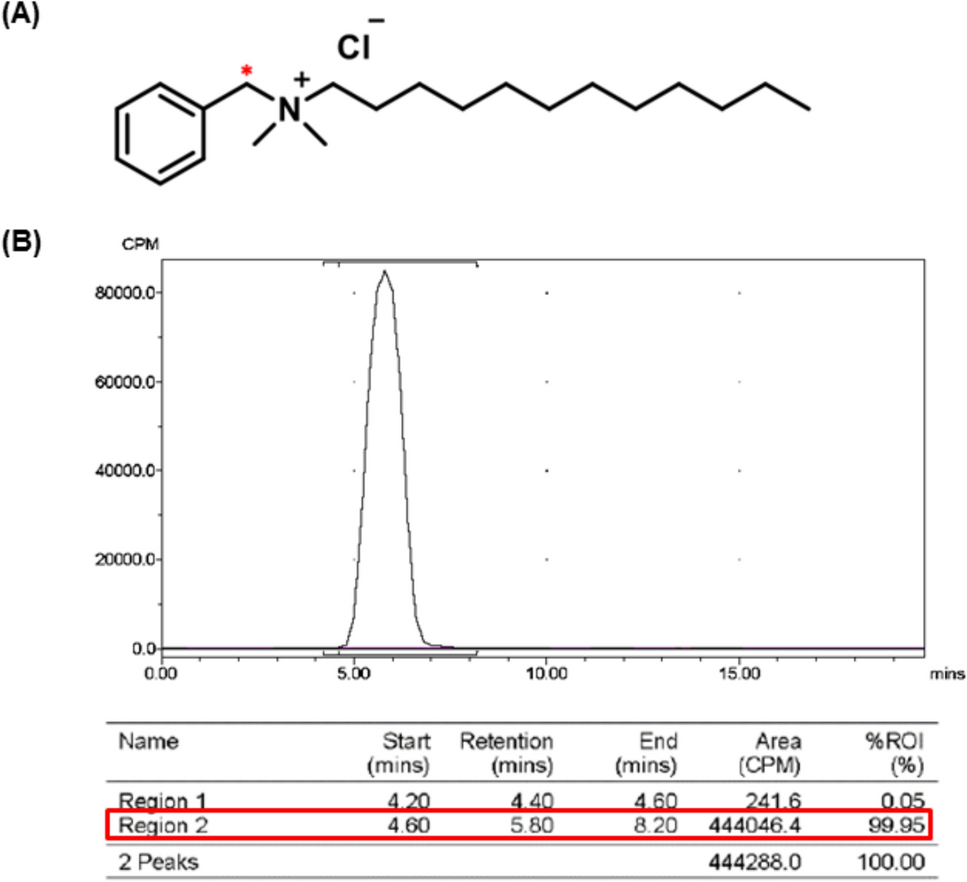
The radiolabeled test substances were [14C]-labeled C12-benzalkonium chloride ([14C]-C12-BKC) and its corresponding non-labeled analog (C12-BKC) were used in this study. The radiolabeled compound was previously synthesized and characterized as described in earlier studies [11, 19], and the position of the [14C]-radiolabel is indicated by an asterisk in Fig. 1a. The radiochemical purity of the labeled compound was greater than 95%, with a specific activity of 2.04 GBq/mmol, as confirmed by radiochromatographic analysis (Fig. 1b).
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Chemical structure and radiochemical characterization of [14C]-labeled C12-benzalkonium chloride ([14C]-C12-BKC). a Chemical structure of C12-benzalkonium chloride, with the [14C] radiolabel indicated by an asterisk (*); b Representative radiochromatogram of [14C]-C12-BKC, demonstrating a radiochemical purity greater than 95%
Non-labeled C12-BKC was purchased from Sigma-Aldrich (St. Louis, MO, USA) and served as an analytical standard to confirm chromatographic retention times and identify the parent compound.
Both labeled and non-labeled compounds were dissolved in sterile distilled water immediately prior to treatment.
Chemicals and reagentsLC–MS grade acetonitrile and water, both containing 0.1% formic acid, were purchased from Fisher Scientific (Hampton, NH, USA). Analytical grade isopropyl alcohol (IPA) was also obtained from Fisher Scientific, while methanol and water were purchased from J.T. Baker (Phillipsburg, NJ, USA).
For radioactivity measurements, Ultima Gold™ liquid scintillation cocktail was purchased from PerkinElmer (Waltham, MA, USA). The scintillator for the flow-through β-RAM detector, FlowLogic U, was obtained from LabLogic (Sheffield, UK). Combustion reagents for the sample oxidizer, Carbo-Sorb E and Permafluor E+ , were supplied by PerkinElmer.
Receptor media and supplements, including Dulbecco’s Modified Eagle’s Medium (DMEM) and penicillin–streptomycin, were purchased from Hyclone (Logan, UT, USA), while fetal bovine serum (FBS) was obtained from Gibco (Grand Island, NY, USA). For the preliminary skin Corrosion/Irritation Test, Phosphate-buffered saline (PBS) and potassium hydroxide (KOH) were obtained from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals used were of analytical grade.
Preparation of ex vivo porcine ear skinPorcine ears were obtained immediately after sacrifice for research purposes from Apures Co. (Pyeong-Taek, Korea). To remove surface contamination, each ear was rinsed sequentially for 1 min with 70% ethanol, DMEM, and warm PBS. Skin discs were excised using a 10 mm biopsy punch (Acuderm, Fort Lauderdale, FL, USA) and temporarily kept in PBS. The prepared skin tissues were positioned epidermis-side up inside 12 mm Millicell inserts (Merck, Cambridge, MA, USA), with a silicone ring affixed to prevent leakage. The inserts were then placed into 6-well plates, and 900 μL of culture medium (DMEM supplemented with 10% FBS and 5% penicillin–streptomycin) was added to the receiver compartment. Prior to the permeation study, the tissues were pre-incubated overnight at 37 °C in a humidified atmosphere containing 5% CO2 to equilibrate the tissue and maintain viability, following the method described by Hwang et al. [20].
Preparation of freeze-killed skinTo prepare metabolically inactive skin (freeze-killed skin), a subset of the freshly excised skin tissues was stored in a deep freezer at −70 °C for at least 2 weeks. Prior to use, the frozen skins were subjected to two freeze–thaw cycles at room temperature for complete abolition of metabolic activity as freezing and storage processes are known to destroy cutaneous enzymatic function [21].
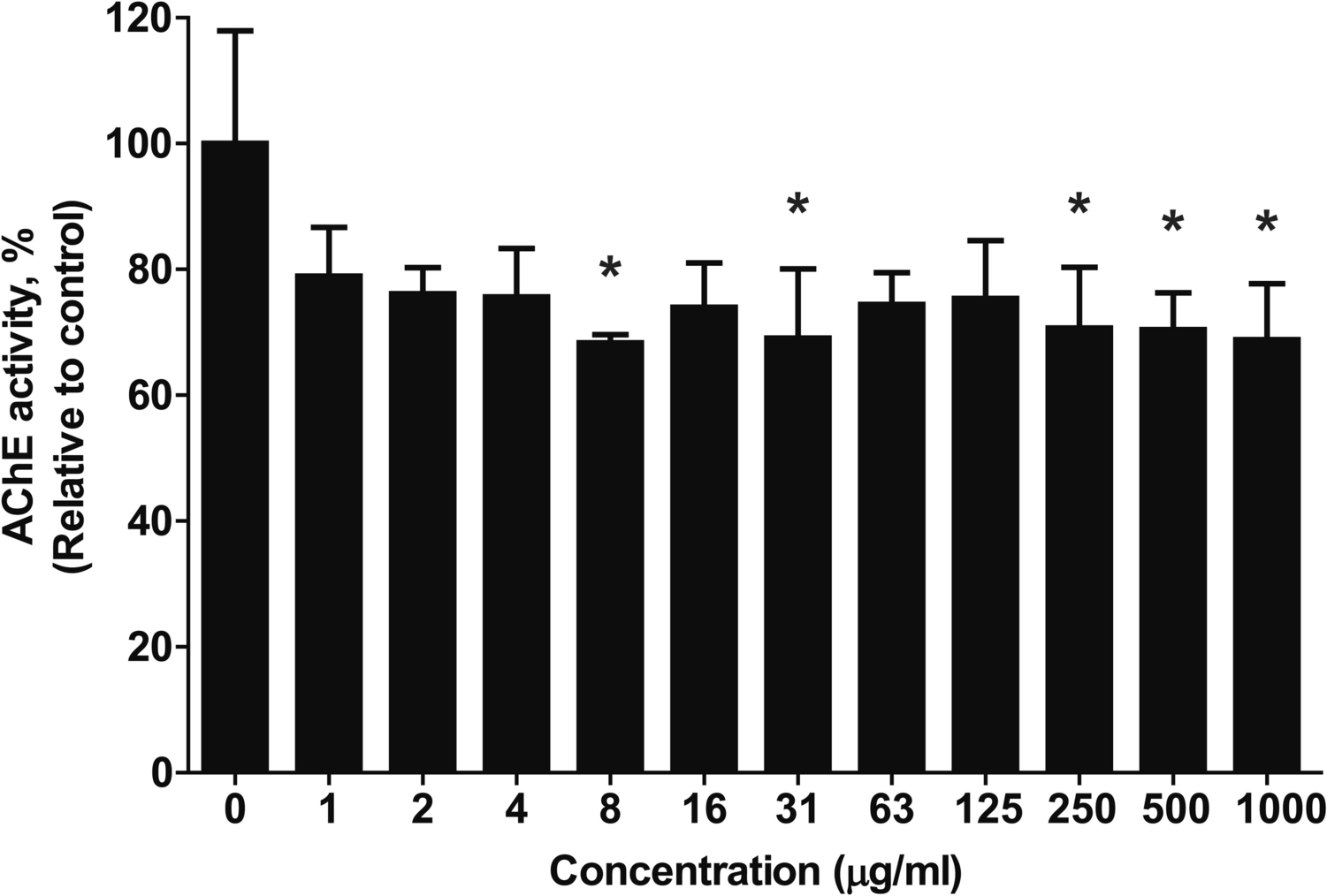
Preliminary skin corrosion/irritation test for dose selectionTo determine the maximum non-toxic dosing concentration for the subsequent permeation study, a preliminary skin corrosion and irritation test was performed using the viable skin tissues. To evaluate concentration-dependent toxicity, 40 µL of C12-BKC solutions at various concentrations (0.01%, 0.1%, and 1%) were topically applied to the epidermal surface (n = 1 per group). The exposure conditions included a 3 min treatment at room temperature and a 60 min treatment at 37 °C, designed to distinguish between corrosive (UN GHS Category 1) and irritant (Category 2) potentials.
Following exposure, the tissues were rinsed with PBS to remove residual test substances and post-incubated for 40 ± 2 h. Skin viability was then quantified using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) assay. A 300 µL aliquot of CCK-8 solution (diluted 1:20 in serum-free medium) was added to each well, and tissues were incubated for 2.5 h at 37 °C. The optical density (OD) was measured at 450 nm using a microplate reader, and cell viability was calculated relative normalized by tissue weight. 8 N KOH was utilized as positive control and distilled water as negative control. Based on the viability cut-off criteria (Corrosive: < 25% viability; Irritant: < 50% viability), the final dosing concentration for the metabolic study was selected.
Treatment and experimental designTopical application was performed by applying 40 µL of the [14C]-C12-BKC dosing solution to the epidermal surface of each skin disc (approximately 51 µL/cm2). Although OECD Test Guideline 428 recommends a finite dose application (typically 10 µL/cm2), a higher application volume and radioactivity dose were employed in this study. This adjustment was made to ensure sufficient radiometric detection sensitivity for the identification of trace metabolites and low-level permeants, considering the anticipated low percutaneous absorption rate of the test compound.
The actual concentration of the dosing solution was determined to be 261.60 µg/mL with a radioactive concentration of 42.48 µCi/mL by LSC analysis. This concentration was selected based on a preliminary skin corrosion and irritation assay described above, ensuring that the applied dose remained within the non-irritant range (< 300 ppm) to preserve skin barrier integrity (viability data presented in the Results section). Consequently, the applied dose was maintained below the determined toxicity threshold to preserve skin viability and ensure the physiological relevance of the metabolic study.
The treated tissues were incubated at 37 °C in a humidified atmosphere containing 5% CO2. The receptor compartment was filled with 0.9 mL of receptor medium, consisting of DMEM supplemented with 10% FBS and 5% penicillin–streptomycin.
Experiments were terminated at predetermined time points: 0, 1, 4, 8, 12, and 24 h. For each time point, three experimental groups were established (n = 3 per group) to compare metabolic profiles and permeation characteristics: Vehicle Control (Viable skin treated with sterile distilled water), Viable Skin Group (Freshly excised skin treated with the [14C]-C12-BKC solution), Freeze-killed Skin Group (Metabolically inactivated skin treated with the [14C]-C12-BKC solution).
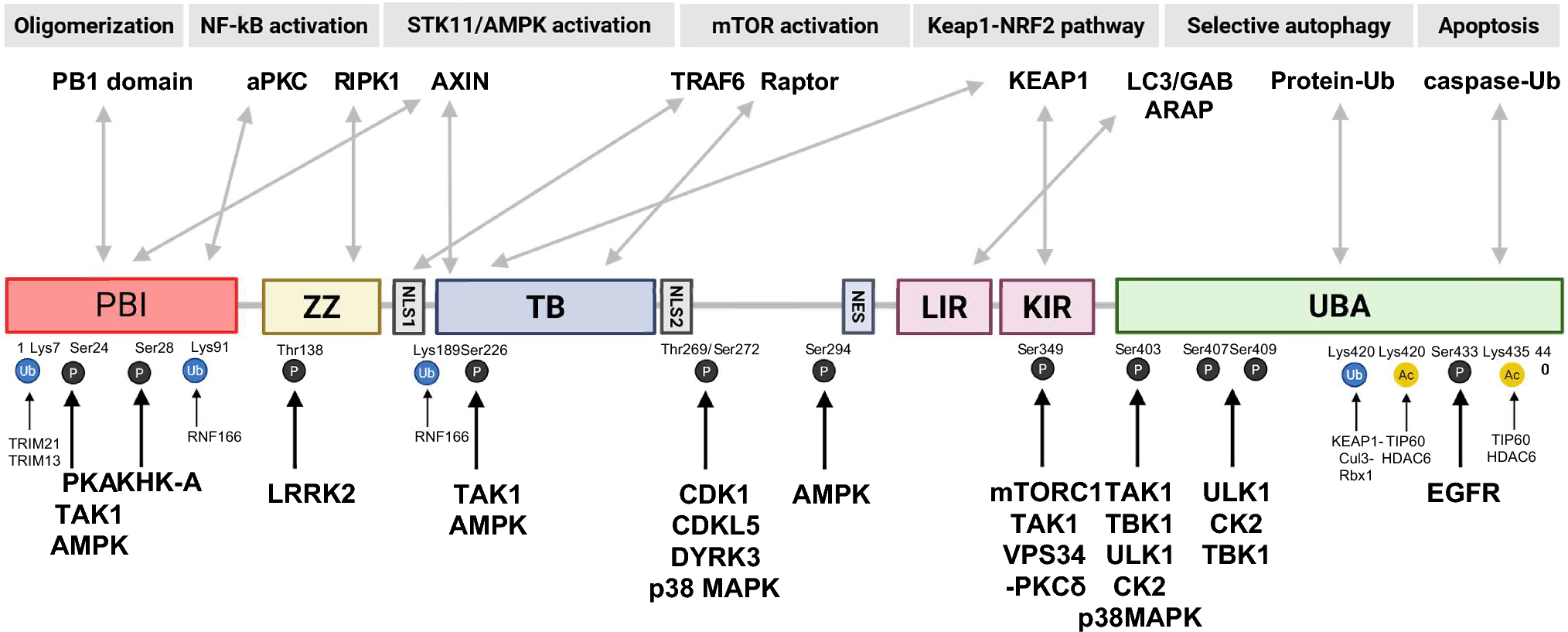
Sampling and extraction proceduresFor the analysis of skin tissue, a two-step extraction method was employed using optimized solvent systems based on a previously reported metabolome extraction strategy [22] to maximize the recovery of the test substance and its metabolites. A schematic overview of the sequential extraction procedure is provided in Fig. 2.
Fig. 2 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Schematic workflow of the sequential two-step extraction procedure for isolating [14C]-C12-BKC and its potential metabolites from porcine skin tissues. Step 1 involves bead homogenization in 80% methanol, and Step 2 involves secondary extraction of the remaining pellet with 100% isopropanol
Specifically, the exposed skin area was excised and immediately transferred into bead tubes prefilled with 300 μL of cold 80% methanol. To instantly terminate metabolic activity, the tubes were stored in a deep freezer for at least 30 min. The tissues were then homogenized using a bead homogenizer (Lysera, Biotage). Following homogenization, distilled water was added to the extract to adjust the final solvent composition to 50% methanol for compatibility with UPLC analysis. The mixture was vortexed and centrifuged (13,500 rpm, 4 °C, 10 min), and the supernatant was collected as the primary extract (from 80% MeOH).
Subsequently, the remaining tissue pellets were subjected to secondary extraction to recover any entrapped residues. 100% isopropanol (IPA) was added to the pellets, and the mixture was vortexed at 4 °C. Distilled water was then added to adjust the composition to 50% IPA prior to analysis. After centrifugation (13,500 rpm, 4 °C, 10 min), the supernatant was collected as the secondary extract (from 100% IPA). The final residual tissue pellets were collected for LSC analysis using a tissue oxidizer.
Preliminary validation studies confirmed that this sequential extraction method (80% MeOH followed by 100% IPA) significantly improved the extraction efficiency compared to using 50% methanol alone. As summarized in Table 1, the addition of the secondary IPA extraction step increased the total recovery to 85.3 ± 10.7%.
Table 1 Extraction efficiency of [14C]-C12-BKC from porcine skin tissues using optimized solvent systemsFor the receptor medium, samples were mixed 1:1 (v/v) with methanol to precipitate proteins, centrifuged (13,500 rpm, 4 °C, 10 min), and the supernatants were collected for analysis.
Radioactivity measurementMeasurement of total radioactivity in all samples was performed using a liquid scintillation counter (LSC; Tri-Carb 5110TR, PerkinElmer). For liquid samples, including surface washing solutions, primary and secondary tissue extracts, and receptor medium, aliquots were mixed directly with Ultima Gold™ scintillation cocktail and analyzed by LSC.
The residual tissue pellets remaining after the secondary extraction were combusted using a sample oxidizer (Model 307, PerkinElmer). The evolved [14C]-CO2 was trapped in Carbo-Sorb E and subsequently mixed with Permafluor E+ scintillator to ensure complete recovery of non-extractable radioactivity. The recovery efficiency of the oxidizer was verified using [14C]-standard spiked control samples, and the measured radioactivity was corrected accordingly.
To ensure measurement accuracy and statistical reliability, radioactivity was measured for 10 min in triplicate for each sample. The average raw counts per minute (CPM) were converted to disintegrations per minute (DPM) using automatic quench correction curves.
UPLC–UV and β-RAM method for quantificationChromatographic separation and quantification of [14C]-C12-BKC and its metabolites were performed using a Waters ACQUITY UPLC I-Class system (Waters) coupled to a β-RAM 5 radioactivity detector (LabLogic). Separation was achieved on a Waters ACQUITY UPLC CSH C18 column (2.1 × 100 mm, 1.7 μm) maintained at 38 °C. The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. The flow rate was set at 0.30 mL/min with an injection volume of 20 μL. The detailed instrumental conditions and the gradient elution program are summarized in Table 2. The total run time was 35 min, including a re-equilibration step to restore starting conditions. UV chromatograms were recorded at 215 nm to simultaneously assess C12-BKC peaks and assist in peak identification.
Table 2 Optimized UPLC-UV and β-RAM settings for sensitive quantification of [14C]-C12-BKC and its metabolitesRadioactivity was detected using the β-RAM 5 detector equipped with a customized 100 μL liquid scintillation flow cell. This reduced flow cell volume was specifically selected to minimize on-flow peak broadening and maintain UPLC resolution, in accordance with optimized radioactivity detection strategies described by Cuyckens et al. [23]. The column effluent passed through the UV detector before being mixed with FlowLogic U scintillation cocktail, delivered via a separate pump at a flow rate of 0.6 mL/min. Due to the serial configuration of the detectors, a constant time offset of 20 s was applied to precisely align the UV and radio traces. Data acquisition and integration were performed using Laura software (version 4.2.37, LabLogic) for radiochromatograms and MassLynx software (version 4.2, Waters) for UV data.
Linearity of the detector response was evaluated using standard solutions of [14C]-C12-BKC. The calibration curve was linear over the range of 0.2–100 μg/mL for UV detection and 0.01–2.0 μCi/mL for radiochemical detection, with correlation coefficients (R2) exceeding 0.999.
Metabolic profiling was performed by separately analyzing aliquots from skin surface washes, skin extracts, and receptor media. The total radioactivity corresponding to the parent drug and potential metabolites was calculated by summing the absolute amounts detected in each compartment. The chromatographic retention time of the parent compound was confirmed using the non-labeled and radiolabeled standards prior to sample analysis.
StatisticsAll experimental data were presented as the mean ± standard deviation (S.D.) of at least three independent replicates (n ≥ 3). Data processing, calculation of mass balance recovery, and visualization of graphs were performed using Microsoft Excel (Microsoft Corporation). Statistical comparisons between viable and freeze-killed skin were conducted using independent two-tailed Student’s t-tests to assess differences at corresponding time points. All statistical analyses were performed using IBM SPSS Statistics software (Version 23.0, IBM Corp.). Differences were considered statistically significant at p < 0.05.
Comments (0)