
Remember me


This trial was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice (ICH-GCP) guidelines and the ethical principles outlined in the Declaration of Helsinki. The study protocol was approved by the Advarra Institutional Review Board, 6100 Merriweather Dr., Suite 600, Columbia, Maryland 21044, (Institutional Review Board (IRB) case number Pro00080984) prior to participant screening. Written informed consent was obtained from each participant before initiation of any trial-related procedures.
2.2 Investigational ProductsThe investigational products included the test product (TP), CBtru®, and the reference product (RP), Epidyolex®. CBtru® (TP) is manufactured by emulsification of an inner oil phase (containing CBD) into an aqueous outer phase and subsequently spray-dried, sieved, and packed in alubags. CBtru® was then filled into hard-shell, gelatin capsules containing 66.68 mg of CBD each, along with standard excipients. Epidyolex® (RP), a commercially available CBD formulation, was sourced by dsm-firmenich (Kaiseraugst, Switzerland). It was provided as an oily oral solution containing 100 mg/mL CBD, with sesame seed oil, dehydrated alcohol, strawberry flavor, and sucralose as inactive ingredients. Both products were manufactured under Good Manufacturing Practice (GMP) conditions. Each administered dose delivered 400 mg of CBD: the TP as six capsules per dose, and the RP as 4 mL of solution per dose.
2.3 SubjectsHealthy adult male and female participants, aged 19 to 55 years, with a body mass index (BMI) between 18.0 and 29.9 kg/m2 and body weight ≥ 50 kg, were eligible for inclusion. Participants were naïve or light recreational users of oral or inhaled cannabis or hemp (averaging fewer than two uses per month). Additionally, participants were not habitual users of nicotine products, having abstained from all nicotine use for more than three months prior to the PK assessment. Female participants were either of non-childbearing potential or confirmed to be non-pregnant and non-lactating at each study visit.
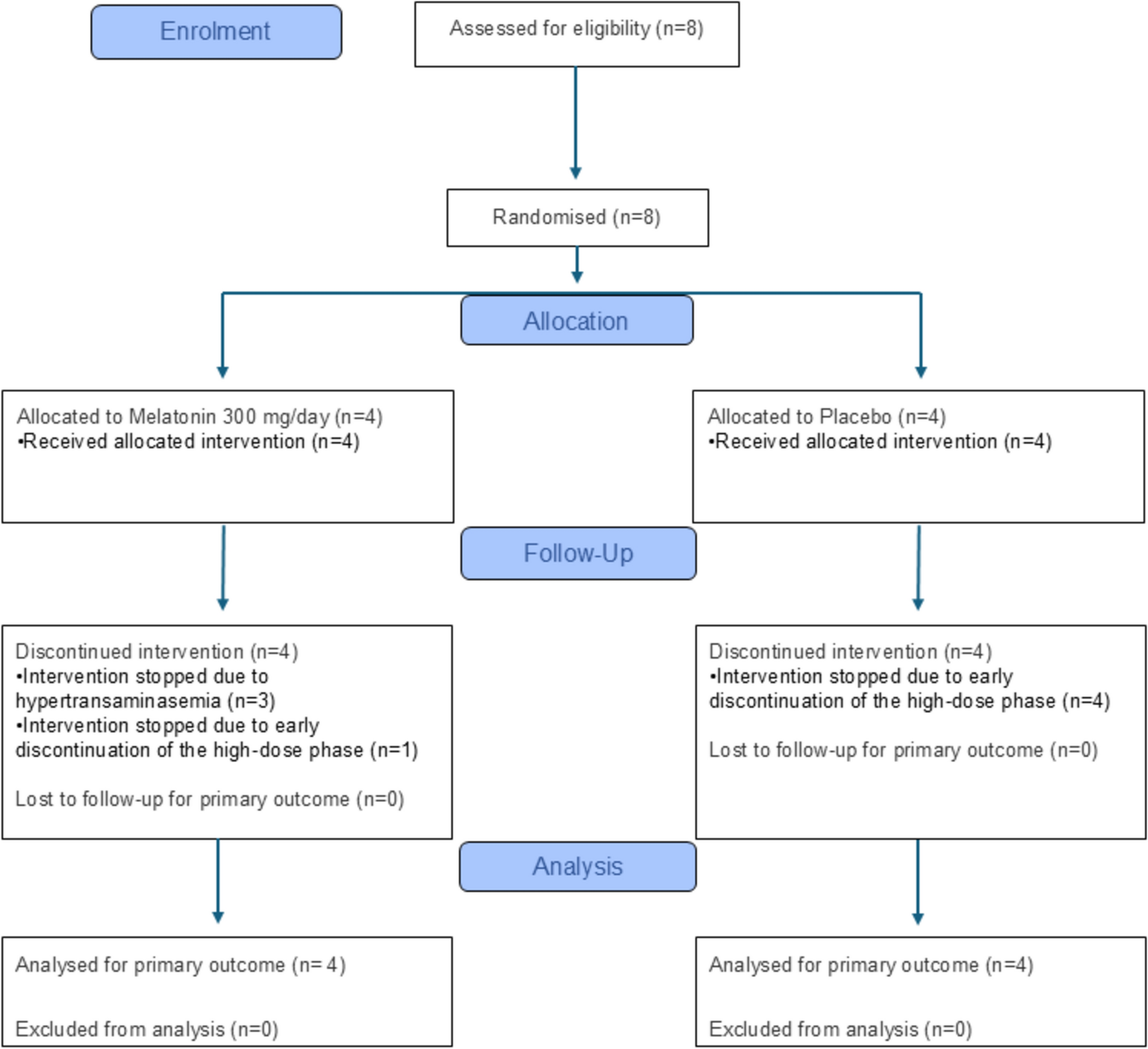
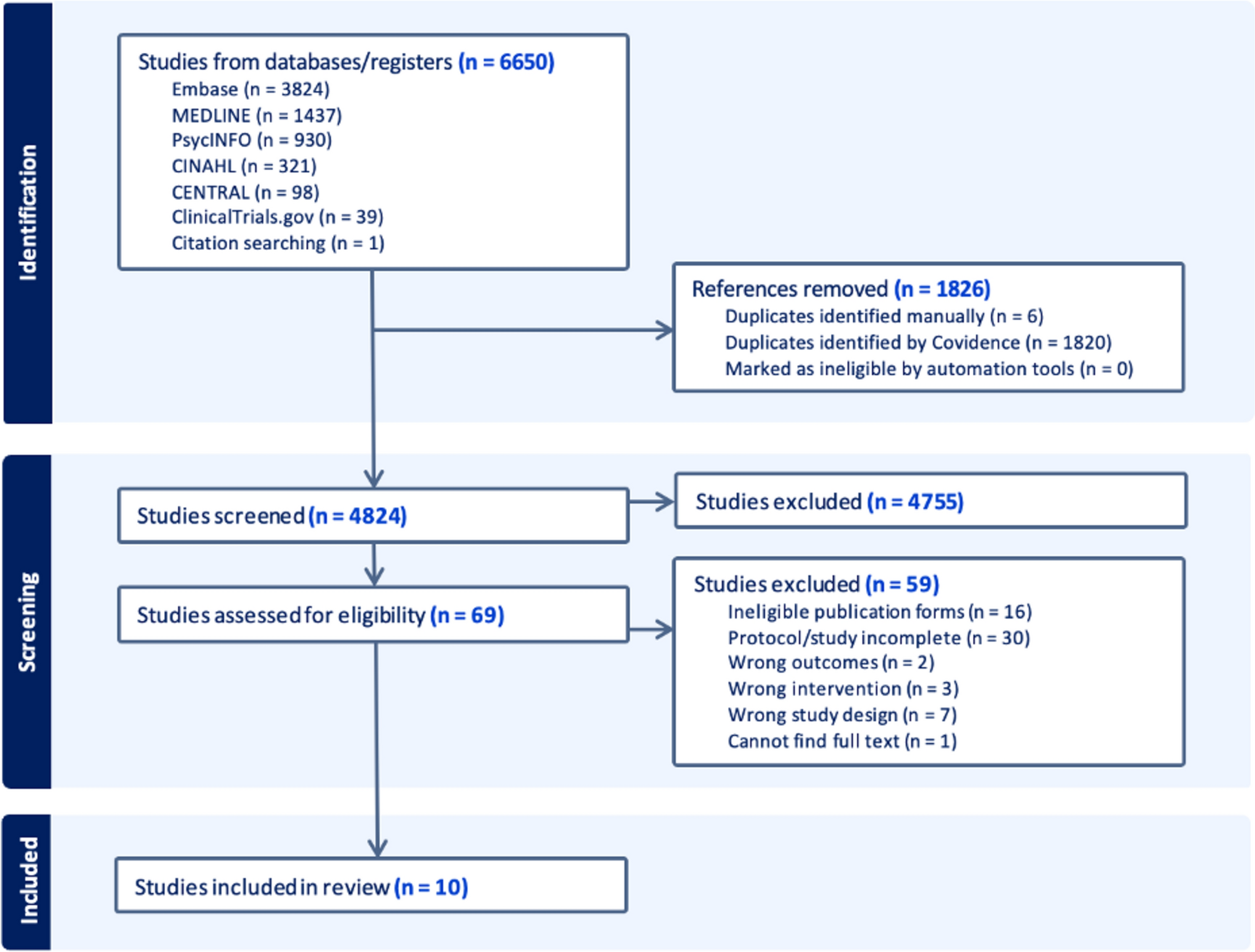
2.4 Trial DesignThis randomized, open-label, four-period crossover study was conducted at a single site in Canada from August 30 to December 16, 2024. The study was registered on ClinicalTrials.gov (NCT06578455). Thirty-two participants (16 males and 16 females) were randomized equally (1:1:1:1) to one of four treatment sequences involving single-dose administration of CBtru® (TP) and Epidyolex® (RP) under both fed and fasted conditions (Fig. 1). For each of the four sequences, the first two periods were conducted in the fed state, with the fasted periods administered thereafter. Participants were stratified by sex; 4 males and 4 females were assigned to every sequence.
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Participant disposition. CBtru® the test product, Epidyolex® the reference product, RP reference product, TP test product
In the fasted condition, participants began overnight fasting for at least 10.0 h prior to dosing. Water intake was permitted until 1 hour before administration. In the fed condition, participants began fasting for at least 10.5 h prior to dosing and consumed a standardized high-fat, high-calorie breakfast (800–1000 kcal, ~ 50% from fat) within 30 min before dosing. The meal included two eggs cooked in butter, two strips of bacon, two slices of toast with butter, 113 g of hash browns, and 240 mL of whole milk, as recommended by regulatory guidelines for food-effect studies. All meals and dosing events were supervised by clinical staff to ensure compliance.
Each study product was administered at a 400-mg dose on Day 1 of each treatment period. A minimum 14-day washout separated each period. Each treatment period included two in-clinic visits, i.e., one full day at the clinic and a short follow-up visit the morning after, resulting in a total of nine visits overall: Screening (Visit 1), Day 1 (Visits 2, 4, 6, 8), and Day 2 post-dose (Visits 3, 5, 7, 9). Participants received a follow-up phone call 14 to 28 days after the last dose of the study product to review any changes in health status and the use of concomitant treatments. For each participant, the timing of this call corresponded to their longest washout period plus one day during the study. The study duration for each participant was approximately 8–20 weeks.
2.5 Determination of Sample SizeThe Health Canada guidance for comparative bioavailability studies recommends a minimum of 12 participants [13]. To accommodate potential sex differences and participant attrition, 32 participants (16 males, 16 females) were enrolled. Sample size estimates were guided by data from Taylor et al. [14], in which area-under-the-curve (AUC) values from 12 participants dosed with 1500 mg CBD under fed and fasted conditions were analyzed. A ~ 4:1 ratio of fed to fasted AUC was observed, with a 90% confidence interval (CI) of ± 20%. With 32 instead of 12 participants in this study, the CI was expected to narrow from ± 20% to ± 12%, whereas the 400-mg dose instead of the 1500-mg dose in this study was expected to widen the CI from ± 12% back to ± 20%.
2.6 Blood Collection and Plasma Sample PreparationBlood samples were collected via intravenous catheter or venipuncture and centrifuged at 1200 RCF for 10 minutes at 15–20 °C. Plasma was stored at − 80°C (± 20 °C). Pharmacokinetic sampling occurred at each in-clinic visit during the four treatment periods (Visits 2–9) and included analyses of CBD, 7-OH-CBD, and 7-COOH-CBD. Fourteen samples were collected per period, including a pre-dose baseline sample (within 90 minutes prior to dosing [fasted] or prior to starting the high-fat, high-calorie breakfast [fed]). Post-dose samples were collected at 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0, 12.0, and 24.0 hours. Actual timepoints were recorded and used for PK analyses.
2.7 BioanalysisA total of 1708 plasma samples were analyzed, with 35 (2.05%) identified as potentially hemolyzed. Bioanalyses were performed at the dsm-firmenich Bioanalytics lab (Kaiseraugst, Switzerland) using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. Samples were diluted with buffer and spiked with internal standards (d3-CBD, d3-7-OH-CBD, d3-7-COOH-CBD) that were purchased at Sigma-Aldrich Chemie GmbH, Buchs, Switzerland, before liquid-liquid extraction using isobutanol-n-hexane and centrifugation. Samples were analyzed with no preceding β-glucuronidase hydrolysis. After evaporation of the organic phase, residues were reconstituted in methanol-water and injected into the LC-MS/MS system (Sciex 7500 TQ) equipped with an ESI (electrospray ionization) source set to negative ion mode. Detection was achieved using MRM transitions: CBD (m/z = 313 → 245), 7-OH-CBD (m/z = 329 → 268), and 7-COOH-CBD (m/z = 343 → 179). Quantification was based on external calibration. Accuracy and precision were monitored via daily analysis of control samples. The lower limit of quantification (LLOQ) was 1.00 ng/mL, 0.50 ng/mL, and 5.00 ng/mL for CBD, 7-OH-CBD, and 7-COOH-CBD, respectively.
2.8 Pharmacokinetic (PK) AssessmentPharmacokinetic parameters were estimated using a non-compartmental model and SAS version 9.4. Evaluated parameters included:
AUC0–24: area under the plasma concentration-time curve from time 0 to 24 h.
AUCinf: area under the concentration-time curve from time zero to infinity.
Cmax: maximum concentration in plasma.
tmax: time to reach maximum (peak) plasma concentration (Cmax).
t1/2: elimination half-life.
AUC0–24/AUCinf: ratio of partial to total exposure.
CL/F: oral clearance of the drug from plasma
Vz/F: apparent volume of distribution.
MRAUC0–24: metabolite-to-parent ratios for AUC0–24.
MRCmax: metabolite-to-parent ratios for Cmax.
For summarizing PK concentrations and plotting, all concentrations below the lower limit of quantification (BLQ) were set to zero, except when a BLQ value occurred between two non-BLQ concentrations, in which case it was set to missing. When calculating AUC, BLQ values occurring before the first measurable concentration were set to zero, while all BLQ values after the first measurable concentration were set to missing.
2.9 Safety AssessmentsSafety was monitored by assessing adverse events (AEs), 12-lead electrocardiograms (ECGs), vital signs, clinical laboratory parameters, and abbreviated physical examinations. Adverse events were documented from the time of informed consent through the follow-up call. Electrocardiograms were recorded pre- and post-dose during dosing visits. Vital signs (blood pressure, heart rate, respiratory rate, temperature) were measured seated at all visits. Blood samples for hematology and chemistry were collected pre-dose at Visits 1, 2, 4, 6, 8, and 12 h post-dose at Visits 2, 4, 6, and 8. Fasting samples were used where applicable. Abnormal lab values were flagged by the central lab as ‘high’ or ‘low’ based on reference ranges. A serious AE was defined as an AE that resulted in death, was life-threatening, required hospitalization, or caused significant disability. Abbreviated physical examinations were performed at key visits, and symptom-driven assessments were conducted as needed. Safety data were reviewed after each treatment period by the central medical monitor according to the study’s safety management plan.
2.10 Statistical AnalysesStatistical analyses were performed using SAS version 9.4. Descriptive statistics were calculated for continuous variables (e.g., mean, standard deviation [SD], median, minimum, maximum). For PK parameters (e.g., Cmax, AUC0–24, AUCinf, CL/F, Vz/F), geometric means and coefficients of variation (CV%) were also computed. For categorical (qualitative) variables, frequency tables showing the counts and proportions were generated. Significance level was set at 0.05.
2.10.1 Primary, Secondary, and Exploratory Endpoint Analysis (PK Parameters)The PK and safety objectives were assessed under both fasted and fed conditions. The primary objective was to evaluate the AUC0–24 of CBD of the test product (TP) versus the reference product (RP), in healthy adults. The secondary objectives included the assessment of additional PK parameters AUC0–24, Cmax, tmax, t1/2, AUCinf, and AUC0–24/AUCinf for CBD and the CBD metabolites 7-OH-CBD and 7-COOH-CBD, and TDE (“total drug exposure” as sum of AUC0–24 for CBD, 7-OH-CBD, and 7-COOH-CBD) of the TP versus the RP. The exploratory objectives included the assessment of PK parameters AUC0–24, Cmax, tmax, t1/2, AUCinf, and AUC0–24/AUCinf for CBD, 7-OH-CBD, 7-COOH-CBD, and TDE of each study product between fasted and fed conditions.
Primary and secondary endpoints were centered on comparisons between TP and RP, while exploratory endpoints assessed differences between fasted and fed conditions. To compare TP and RP under each condition, an ANOVA was performed on log-transformed values of AUC0–24, AUCinf, and Cmax in line with pharmacokinetics guidance documents (e.g., FDA). The model included treatment sequence, treatment, and period as fixed effects, with subject nested within sequence as a random effect. Body weight was treated as a covariate. Treatments consisted of the following four combinations: TP-fed, TP-fasted, RP-fed, and RP-fasted.
Least-squares means (LSM), differences in adjusted treatment means, and associated standard errors (SEs) were calculated. Geometric least square mean ratios (GLSMRs) and their 90% CIs were derived for TP-fed versus RP-fed and TP-fasted versus RP-fasted. For exploratory endpoints, GLSMRs, and 90% CIs were similarly calculated for TP-fed versus TP-fasted and RP-fed versus RP-fasted using the same model. Tmax comparisons were conducted using a non-parametric Wilcoxon test.
Exploratory PK parameters MRAUC0–24, MRCmax, CL/F, and Vz/F were calculated using non-compartmental analysis. The MR assesses relative metabolite exposure, CL/F reflects drug elimination efficiency accounting for oral bioavailability, and Vz/F estimates the extent of systemic drug distribution. Additional exploratory evaluations included the active drug exposure (sum of AUC0–24 for CBD and 7-OH-CBD) of each study product, which was compared between TP and RP and between fasted and fed conditions.
2.10.2 Safety AnalysesThe safety objective was to evaluate the tolerability of TP and RP in healthy adults. Adverse events were coded according to the contract research organization's (CRO's) medical dictionary. The safety set included all randomized participants who received at least one dose of the study product. Adverse events occurring on or after the first dosing of each period, or worsening in severity/frequency post-dosing, were considered treatment-emergent adverse events (TEAEs) and summarized by term, severity, and relationship. Each participant contributed once per TEAE incidence, regardless of the number of occurrences.
Comments (0)