
Remember me

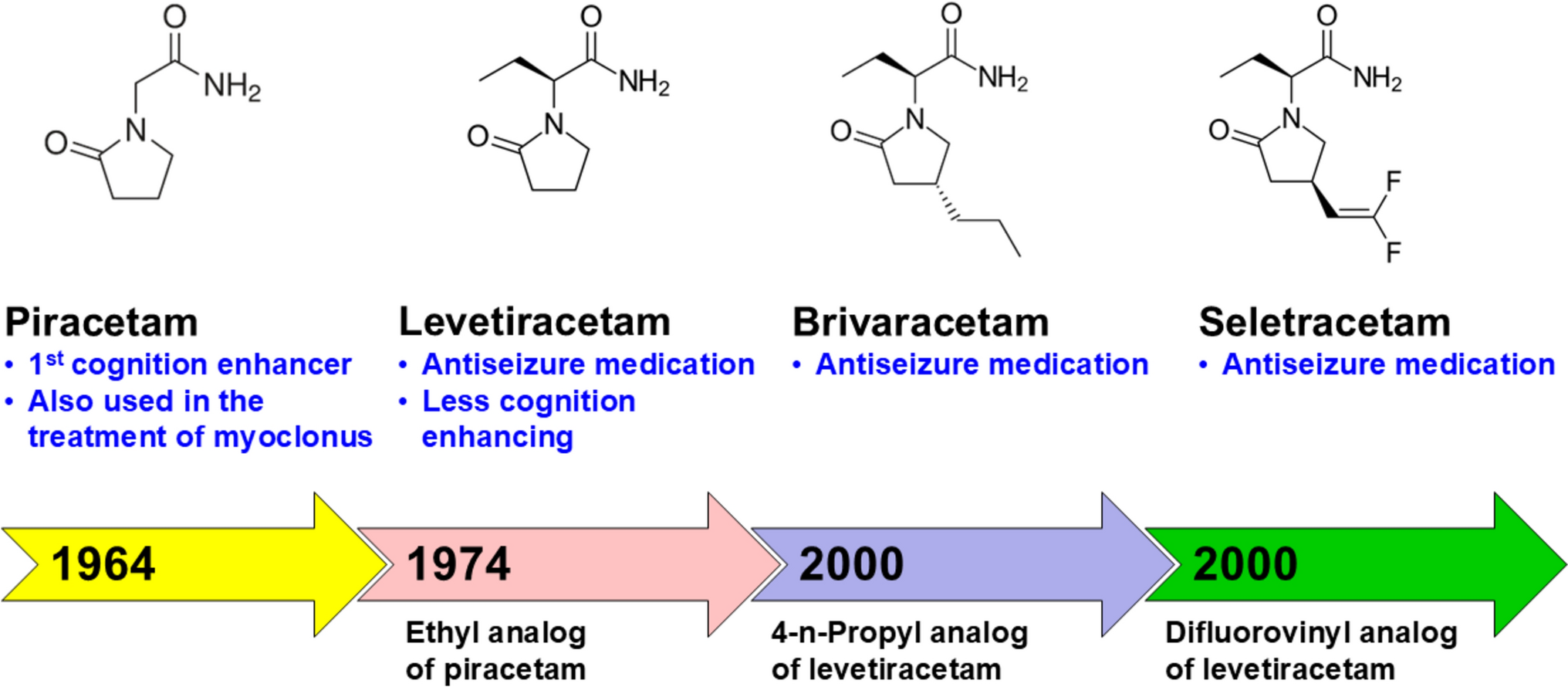

After developing the ASMs levetiracetam (LEV) and brivaracetam (BRV), which both act mainly by modulation of SV2A, UCB Pharma thought about the feasibility of designing a molecule with both presynaptic and postsynaptic activity via interaction with SV2 proteins and the GABAA receptor, respectively [12]. The rationale for targeting these proteins was based on observations in the early 2000s that LEV markedly potentiated the activity of ASMs acting via GABAergic transmission, notably BDZs, in several animal models, resulting in an improved efficacy/safety ratio [51]. Figure 3 illustrates the potentiation of several BDZs by a low subeffective dose (5.5 mg/kg) of LEV in the DBA/2 mouse audiogenic seizure model (for comparison, the ED50 of LEV in this model is 30 mg/kg; Table 1). BDZs act as PAMs of GABAA receptors by binding to the BDZ binding site of the GABAA receptor chloride ionophore complex [52]. As shown in Fig. 3, the full agonists at this site (clonazepam, diazepam, and chlordiazepoxide) and the partial agonist bretazenil were markedly potentiated by pretreatment with LEV, resulting in 11–23-fold increases in anticonvulsant potencies. A similar synergistic interaction between LEV and BDZs was reported for several other seizure and SE models [51]. Importantly, these combinations were not associated with exacerbation of side effects or pharmacokinetic interactions. The anticonvulsant potency of several drugs inhibiting sodium channels (i.e., CBZ or PHT) or calcium channels (flunarizine) was enhanced by LEV to a much lesser degree [51].
Fig. 3 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Potentiation of the antiseizure effect of benzodiazepines (BDZs) by a low dose (5.5 mg/kg) of levetiracetam (LEV) in audiogenic seizure-prone DBA/2 mice. Clonic seizures were used as an endpoint in this model. Anticonvulsant ED50 values, as well as the change in potency by pretreatment with LEV, are illustrated. LEV was administered 60 min before the BDZs. Data are from Kaminski et al. [51]. ED50 median effective dose
3.1 Preclinical Development of PadsevonilThe focus of the subsequent rational medicinal chemistry design program performed by UCB Pharma was to develop a single molecule that could target SV2 proteins with high affinity and the BDZ binding site of postsynaptic GABAA receptors with lower affinity [53]. Low-to-moderate affinity for this site, coupled with a partial agonist profile, could minimize the potential for sedation and respiratory adverse effects and the development of tolerance and dependence, phenomena known to occur with most BDZs [54, 55]. Lead optimization efforts led to the discovery of padsevonil, an imidazothiadiazole heterocycle coupled to a pyrrolidone moiety (Fig. 1).
Padsevonil’s affinity for recombinant human SV2A (pKi 8.5) is 2000-fold greater than that of LEV (pKi 5.2) and 80-fold greater than that of BRV (pKi 6.6), respectively [53]. Padsevonil’s interaction with SV2A differed from that of LEV and BRV, i.e., it exhibited slower binding kinetics with a dissociation half-life of 30 min compared with <0.5 min for LEV and BRV, translating into longer target occupancy. Unlike LEV and BRV, padsevonil also displayed high affinity for the SV2B and SV2C isoforms of SV2 (pKi 7.9 and 8.5, respectively). Like SV2A, the other two SV2 isoforms—SV2B and SV2C—are integral membrane glycoproteins that are present in secretory vesicles of neurons and endocrine cells [56]. The precise function of the SV2 proteins remains elusive; however, current evidence suggests that they play a role in vesicle exocytosis and, thus, the release of neurotransmitters [57, 58].
Recently, the cryo-electron microscopy structures of SV2A in complexes with drugs, including LEV, BRV, and padsevonil, have been reported, indicating a central drug-binding cavity that faces the vesicle interior [59]. Padsevonil differs from racetams such as LEV and BRV in that it occupies both the central cavity (the orthosteric site) and a secondary distinct ligand-binding site (the allosteric site), functionally precluding modulation of SV2A by allosteric modulators such as UCB1244283 [60].
At recombinant GABAA receptors, padsevonil displayed low-to-moderate affinity (pIC50 ≤ 6.1) for the BDZ site. In electrophysiological studies, its relative efficacy compared with the full-agonist reference drug zolpidem was 40%, indicating partial agonist properties [53, 61]. In in vivo receptor occupancy studies in mice, padsevonil exhibited SV2A occupancy at low ED50 (0.2 mg/kg) and BDZ site occupancy at higher doses (ED50 36 mg/kg), supporting in vitro results [53]. Profiling studies confirmed high selectivity of padsevonil for its targets, excluding off-target effects at a wide variety of ion channels, receptors, transporters, and enzymes. Overall, these observations confirm the unique target profile of padsevonil that should translate into combined presynaptic SV2 modulation of neurotransmitter release and postsynaptic potentiation of GABAergic inhibitory transmission.
In proof-of-concept (POC) experiments in the 6-Hz (44 mA) mouse model of difficult-to-treat focal seizures [13], the antiseizure effect of padsevonil was compared with that of LEV and diazepam alone, and with the combinations of LEV with diazepam at doses expected to provide similar occupancy at SV2A (35%) or the BDZ site (2%) (Fig. 4A). Concerning the occupancy data illustrated in Fig. 4A, it is important to note that BDZs such as diazepam exert their anticonvulsants effects at low (~5–15%) GABAA receptor occupancy [62,63,64,65]. However, in the case of partial agonists such as bretazenil, higher receptor occupancy (>60%) is needed for anticonvulsant effects [66]. In comparison, LEV and BRV require 80% SV2A occupancy for antiseizure activity in animal models [67]. Low-level occupancy was selected for the experiments illustrated in Fig. 4 to differentiate the activity of padsevonil [13]. Protection from 6-Hz seizures offered by padsevonil, even at only 35% SV2A occupancy, was almost 70% and significantly greater than that provided by LEV in combination with diazepam (Fig. 4B). These observations suggest that padsevonil’s antiseizure properties are due to a differentiated mode of action that provides greater protection than co-administration of an SV2A ligand and a BDZ. The interaction of padsevonil with SV2B and SV2C may contribute to the enhanced antiseizure effects. Unfortunately, it was not assessed whether a BDZ antagonist such as flumazenil would decrease the anticonvulsant efficacy of padsevonil. Without such an experiment, the POC data shown in Fig. 4 do not prove that 2% GABAA receptor occupancy with only partial agonism contributed to the antiseizure effect of padsevonil.
Fig. 4 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Antiseizure effect of padsevonil (PSL; 0.17 mg/kg) in the 6-Hz mouse model of focal seizures compared with that of levetiracetam (LEV; 1.83 mg/kg) and diazepam (DZP; 0.017 mg/kg), as well as the combination of DZP with LEV. As shown in A, the drugs were administered at doses associated with similar in vivo SV2A (35%) and benzodiazepine (BDZ) site (2%) occupancies (based on results of in vivo occupancy studies). In B, a significant difference in antiseizure potency between PSL and LEV+DZP is indicated. Data are from Leclercq et al. [13]
In contrast to cenobamate, padsevonil was not tested in the NINDS ETSP program; instead, all preclinical evaluation in seizure models was performed in-house by UCB Pharma. The only exception was the intrahippocampal kainate (IHK) mouse model, which was performed by SynapCell (Grenoble, France), which also performs drug testing in this model for the ETSP [23]. As shown in Table 1, in acute mouse models, padsevonil exerted potent, dose-dependent protection against seizures induced by PTZ, 6-Hz (44 mA), and audiogenic stimulation with ED50s ranging from 0.16–4.8 mg/kg i.p. In this respect, padsevonil was markedly more potent than clinically established ASMs and cenobamate. The only exception was the MES model, in which padsevonil was less potent (ED50 92.8 mg/kg) than cenobamate (9.8 mg/kg). However, in the early 1990s, the MES model failed to detect the anticonvulsant activity of LEV, whereas this drug was highly potent and effective in the rat amygdala kindling model of TLE [68], which correctly predicted the clinical efficacy of this benchmark ASM. Thus, low or absent antiseizure activity in the MES test does not necessarily predict low or absent clinical efficacy, which is also shown by the lack of efficacy of other clinically effective ASMs (e.g., vigabatrin, tiagabine) in the MES test [7].
While cenobamate was also tested in the lithium-pilocarpine SE model in rats (Sect. 2.2), padsevonil’s antiseizure effect was evaluated in the mouse pilocarpine model [13]. Padsevonil’s ED50 in this model was 0.19 mg/kg, while the ED50 of LEV was 7.1 mg/kg, and BRV was ineffective, again demonstrating the high potency of padsevonil.
In the amygdala kindling model of TLE in mice, padsevonil suppressed secondarily generalized convulsive seizures (Racine stage 3–5) with an ED50 of 1.2 mg/kg (Table 1). Unfortunately, the ability of padsevonil to suppress focal (stage 1–2) seizures in kindled animals was not evaluated (or reported). When the afterdischarge duration (ADD) in the EEG recorded from the amygdala was used as an electrographic measure of both focal and secondary generalized seizures, padsevonil significantly reduced the ADD by ~60% only at the highest dose (13.9 mg/kg) tested, indicating a lack of efficacy against focal seizures in this model (see Sect. 4). These data would indicate that, at least in this model, padsevonil prevents seizure spread or generalization but does not block focal firing.
In mice with spontaneous recurrent focal seizures induced by intrahippocampal kainate, a widely used mouse model of mesial TLE, padsevonil suppressed focal hippocampal paroxysmal discharges (HPDs) with an ED50 of about 0.4 mg/kg. At the highest dose tested (4.33 mg/kg), HPDs were completely suppressed [13]. The TD50 of padsevonil in the rotarod test was 11.8 mg/kg, resulting in the highest protective index of all ASMs by far, including cenobamate, shown in Table 1.
As shown in Table 2, padsevonil was also highly effective in the rat amygdala kindling model (ED50 2.4 mg/kg) and in the Genetic Absence Epilepsy Rats from Strasbourg (GAERS) model of spontaneous spike-wave discharges (ED50 0.07 mg/kg). However, as in the amygdala kindling model in mice, only the ED50 for generalized convulsive and not focal seizures was determined in the rat version of the kindling model. Furthermore, no dose-dependent effect on ADD was observed, but ADD was reduced by only about 50% at 2.4, 4.3, and 13.9 mg/kg. As in mice, a high protective index (10.2) was seen in rats. Altogether, these results would suggest efficacy against generalized tonic–clonic but not focal seizures.
In additional experiments using the PTZ seizure threshold test, it was tested whether prolonged treatment with padsevonil leads to loss of anticonvulsant efficacy (i.e., tolerance) [13]. In contrast to diazepam, which is associated with a significant loss of efficacy, no such phenomenon was observed with padsevonil.
More recently, Quinlan et al. [69] reported that padsevonil suppresses PTZ-induced seizures in neonatal rats. While many ASMs, including PB and BDZs, induce neuronal cell death in neonatal rats [70], padsevonil did not. This suggests that padsevonil may offer therapeutic benefit and a favorable safety profile for the treatment of neonatal seizures [69].
3.2 Translation of SV2A and GABAA Receptor Target Engagement by Padsevonil from Mice to HumansThe advantage of the SV2A and GABAA receptor targets of padsevonil is that in vivo receptor occupancy studies can be performed in animals and humans, using highly selective 11C-labeled ligands and positron emission tomography (PET). The nonclinical studies described above showed that padsevonil is highly active in models of drug-resistant epilepsy at doses that lead to >95% SV2A occupancy and <20% occupancy on GABAA receptors. For translating SV2A and GABAA receptor target engagement from rodents to humans, UCB Pharma chose the ED50 of padsevonil (1.2 mg/kg) in the mouse amygdala kindling model, because, as described above, this model (in rats) had correctly predicted the clinical efficacy of LEV. As shown in Fig. 5, in vivo occupancy studies in mice showed that at this dose, padsevonil exhibited >90% SV2A occupancy and ~10% occupancy at GABAA receptors [71]. The differential proportionality of target engagement was retained in human volunteers as shown in PET studies following oral administration of 400 mg padsevonil (Fig. 5). Dose response experiments demonstrated that quasi full saturation at SV2A was already obtained at 100 mg of padsevonil, but simulations projected that SV2A occupancy would be >90% over the entire dosing interval for only 75% of a population receiving padsevonil 100 mg b.i.d. but for 90% of a population receiving padsevonil 300 or 400 mg b.i.d. [71]. On the basis of these data, a dose of 400 mg b.i.d. was chosen for the clinical phase 2a POC trial (see Sect. 3.3). PET studies with repeated administration of 400 mg in healthy volunteers showed that with this dosing schedule high (>90%) SV2A occupancy was sustained whereas the low (~13%) GABAA receptor occupancy was transient, in that at 2 h and 6 h post-dose, occupancy for the average of all regions of interest was ≤5% for most volunteers [71].
Fig. 5 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Translating SV2A and GABAA receptor target engagement of padsevonil from rodents to humans. In the amygdala kindling model of chronic epilepsy in mice, the ED50 (the padsevonil dose that protected 50% of mice against secondary generalized convulsive seizures) was 1.2 mg/kg. In vivo occupancy studies in mice showed that at this dose, padsevonil exhibited >90% SV2A occupancy and ~10% occupancy at GABAA receptors. The differential proportionality of target engagement was retained in humans as shown in PET studies 2 h following oral administration of 400 mg padsevonil: >90% SV2A and ~13% GABAA receptor occupancy, using 11C-UCB-J as an SV2A ligand and 11C-flumazenil as a ligand for the benzodiazepine site of the GABAA receptor. On the basis of these data and data from lower doses of padsevonil, simulations of b.i.d. dosing regimens projected SV2A occupancy to be greater than 90% over the entire dosing interval for 90% of a population when using a 400 mg b.i.d. dosing regimen. Thus, this regimen was selected for the phase 2 proof-of-concept trial, as it was expected to achieve exposure levels similar to those associated with robust efficacy in the amygdala kindling model (see text for details). Data are from Muglia et al. [71] and Henrik Klitgaard. GABA, gamma-aminobutyric acid; SV2A, synaptic vesicle glycoprotein 2A
3.3 Clinical Evaluation of Padsevonil in Patients with Drug-Resistant Focal EpilepsyUCB Pharma’s padsevonil drug development program goal was to make a dent in the treatment of DRE, and padsevonil was engineered specifically for that purpose [6]. For this reason, the phase 2a POC trial targeted patients with severe focal DRE: patients experienced a median of 10 seizures/week, and 75% patients had failed ≥8 ASMs [71]. The innovative POC trial design with maximal signal detection in a small patient population (n = 55) with a short treatment duration is illustrated in Fig. 6. Patients were randomized to receive placebo or padsevonil as an add-on to their stable ASM regimen. After a 3-week inpatient double-blind period, all patients received padsevonil during an 8-week outpatient open-label period. The novel high bar primary endpoint was ≥75% reduction in seizure frequency. As shown in Table 3 and Fig. 7, this primary outcome did not reach statistical significance compared with placebo. Three patients (11.1%) in the placebo group and eight (30.8%) in the padsevonil group experienced ≥75% reduction from baseline in seizure frequency during the inpatient period [odds ratio 4.14 (95% CI 0.90–19.6), P = 0.068]. The secondary outcome, median reduction in weekly focal seizure frequency from baseline during the 2-week inpatient period, was 53.7% in the padsevonil and 12.5% in the placebo groups, a difference of 34.0% (95% CI 3.0–67.5; unadjusted P = 0.026) (Table 3).
Fig. 6 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Design of the randomized, double-blind, placebo-controlled, proof-of-concept (POC) trial to assess the efficacy and tolerability of padsevonil in adults with drug-resistant focal seizures. Adapted from Muglia et al. [71]. aDefined as the last 2 weeks of the inpatient period for the padsevonil (PSL) arm and the first 2 weeks of the inpatient period for the placebo (PBO) arm. bNo more than 3 weeks. cTreatment was initiated at 100 mg/day. dDose could be tapered to 200 mg/kg. OLE, open-label extension
Table 3 Efficacy outcomes of the phase 2a proof-of-concept (POC) trial with a single-dose arm of padsevonil (400 mg b.i.d.) []aFig. 7 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Modified from Van Paesschen et al. [72]
Efficacy outcomes (≥75% responder rates and median percent reductions in seizure frequency) of the phase 2a proof-of-concept (POC) trial with padsevonil [71]. In addition to focal seizures, a post hoc analysis of all seizures was performed, resulting in significant differences between padsevonil and placebo [72]. For details, see text, Table 3, and Fig. 6.
In a post hoc analysis, the incidence of all seizures (focal, generalized, and unclassified) at baseline and during treatment was assessed [72]. In contrast to lack of significant effects on focal seizures, the ≥75% responder rate for all seizures was significantly higher in the padsevonil versus the placebo group (Fig. 7). Furthermore, seizure frequency was significantly reduced by padsevonil. These data thus seem to be consistent with the effects of padsevonil in amygdala kindled rats (Sect. 3.1) in that this drug primarily prevents seizure spread or generalization but is less effective in blocking focal seizures.
Almost half of the patients of the POC trial were concomitantly treated with LEV (n = 20) or BRV (n = 4), which may have affected the efficacy of padsevonil. Therefore, the “SV2A” and “non-SV2A” subgroups were separately analyzed (Table 3). Post hoc analysis of these subgroups indicated significant differences from placebo in the non-SV2A but not the SV2A subgroups. The ≥75% responder rate was 53.3% in the non-SV2A subgroup during the 2-week (only) double blind inpatient phase of the study and 0 in the SV2A subgroup (P = 0.003). Surprisingly, this outcome had not been anticipated, even though prior phase 2–3 RCTs of BRV had shown lesser, statistically nonsignificant efficacy of BRV in patients with LEV as a concomitant medication (see Sect. 3.4.2).
Following the POC trial, UCB started in parallel two large (phase 2b and phase 3) trials [14]. In the dose-finding phase 2b trial (EP0091; NCT03373383; ARISE) of 410 patients, the primary objectives were to characterize the dose–response relationship of padsevonil administered concomitantly with up to three ASMs for seizure reduction in patients with DRE and to evaluate the efficacy of padsevonil (50, 100, 200, 400 mg twice daily) compared with placebo. In the phase 3 efficacy trial (EP0092; NCT03739840; DUET), the primary objective was to evaluate the efficacy of padsevonil (100, 200, 400 mg b.i.d.) administered concomitantly with up to three ASMs compared with placebo for observable focal seizures in patients with DRE. The trial design and patient eligibility were the same for both trials, except for the target doses of padsevonil. Primary efficacy outcomes were the change in log-transformed observable focal seizure frequency from baseline and a 75% responder rate (≥75% reduction in observable focal seizure frequency from baseline) over the 12-week maintenance treatment period.
Both trials failed to reach statistical significance for any padsevonil dose group compared with placebo [14]. In the dose-finding phase 2b trial, numerical improvements were observed for the padsevonil groups compared with placebo (Table 4); however, these did not show a clear dose-dependent response. Seizure freedom during the maintenance treatment phase was achieved in 8.2% at 200 mg and 6.9% at 400 mg of padsevonil versus 0% treated with PBO, respectively, in the dose finding trial, comparable with other ASMs including BRV, lacosamide, and perampanel [10]. In the phase 3 trial, 2.3, 6.8, and 8.3% of patients on padsevonil 100 (n = 44), 200 (n = 44), and 400 mg b.i.d. (n = 36), respectively, versus 4.3% on placebo (n = 46) were seizure free [14].
Table 4 Efficacy outcomes of patients randomized to placebo or padsevonil by randomized dose group for observable focal seizures over the 12-week maintenance period in the phase 2b dose-finding trial (EP0091)[14]aAs in the phase 2a POC trial, a large percentage of patients were on concomitant treatment with LEV or BRV: 54 and 41% in the phase 2b and the phase 3 trials, respectively. A post hoc analysis of the SV2A and non-SV2A subgroups was only provided for the dose-finding phase 2b trial [14]. As shown in Table 4, the 75% responder rate was higher in the non-SV2A group. Overall, 29/149 (19.5%) patients responded with ≥75% seizure suppression in the non-SV2A group compared with 14/175 (8%) patients in the SV2A group, a highly significant difference (P = 0.0020; Fisher’s exact test). These data of the non-SV2A group are illustrated in Fig. 2, demonstrating that the 75% responder rate of this group of the phase 2b padsevonil trial was comparable to the 75% responder rates during 12–13-week-long maintenance periods in pivotal phase 3 or phase 2 double-blind, randomized, placebo-controlled studies of the two most recently approved sodium channel blockers: lacosamide (approved 2009) and eslicarbazepine (2013), and two other recently approved ASMs with different MOAs: BRV (2015) and perampanel (2013). Only cenobamate exhibited markedly higher 75% responder rates. Rademacher et al. [14] concluded that “even without concomitant SV2A drug intake, the efficacy of padsevonil was modest, and 75% responder rates were not better than those observed in pivotal trials with other ASMs in populations of comparable epilepsy severity.” Importantly, the apparently high efficacy of padsevonil observed at 400 mg in the POC trial in a small group of patients (Table 3) was not reproduced in the phase 2 trial in a larger patient group (Table 4; Fig. 2). Thus, the expectation that the rationally developed padsevonil with two synergistic MOAs and high preclinical potency would improve efficacy compared with available ASMs did not come true. Even worse, in patients on chronic treatment with LEV or BRV, padsevonil’s efficacy was clearly below the efficacy of add-on treatment with clinically established ASMs.
3.4 Potential Reasons for the Failure of PadsevonilThe unexpected failure of padsevonil to better the efficacy of other novel ASMs may have several reasons.
3.4.1 Most Animal Models of Seizures or Epilepsy Allow the Determination of Antiseizure Potency but not the Efficacy of Novel CompoundsAs described in Sect. 3.1, padsevonil performed very well in animal models expected to differentiate a drug that would be effective in focal DRE, including intrahippocampal kainate, the amygdala kindled mouse, and the 6-Hz model, in some cases outperforming all existing ASMs. However, the typical approach of ASM testing in animal models primarily focuses on drug potency and not efficacy [73]. Thus, different investigational drugs are compared in terms of their antiseizure ED50s, i.e., the dose suppressing seizures in 50% of the animals, which is calculated from dose–response curves, testing one group of animals per dose. The lower the ED50, the more potent the drug, and high potency is often an important argument for selecting drugs for further development. However, it is the antiseizure efficacy that ultimately determines the clinical usefulness of a new ASM and should be considered during preclinical drug testing. The 6-Hz mouse model of focal seizures is one example of a model that allows determining both the potency and efficacy of drugs. At a high stimulation current (i.e., 44 mA), this test displays a degree of pharmacoresistance to the current standards of care and allows an investigator an opportunity to differentiate their investigational drug on the basis of potency and efficacy by varying the stimulation strength [74]. As shown in Table 1 (see more examples in the review by Löscher and White [26]), increasing the stimulation intensity of the test from 22 to 44 mA reduced the ED50 of most ASMs markedly, whereas the ED50 values of cenobamate and carisbamate were only modestly reduced. Padsevonil was only tested in the 44 mA version of the test,
Comments (0)