Data acquisition
Two microarray datasets (GSE61616 and GSE58720) detailing the gene expression profiles of CIRI and the microarray datasets (GSE106931) detailing the gene expression profiles of macrophage after CIRI were obtained from the Gene Expression Omnibus (GEO) database. The GSE61616 dataset contained five control and five ischemia samples. The GSE61616 samples were from the right hemispheres of male Sprague–Dawley rats that were subjected to 2 h of occlusion followed by 7 days of reperfusion. GSE106931 included sorted CD11b+CD163+ cells from rat brain tissue (from three control rats and three ischemic rats 16 h post-ischemia). GSE58720 contained mouse brain tissue samples from five control and five ischemia samples. The ischemia samples in GSE58720 were obtained from mice subjected to 90 min of MCAO followed by 3 h of reperfusion. Among the three datasets were analyzed with GEO2R. Differentially expressed genes (DEGs) were identified using the cutoff value of adjust. p (adj. p) < 0.05 and |log2 Foldchange|> 1.
Viral particles construction
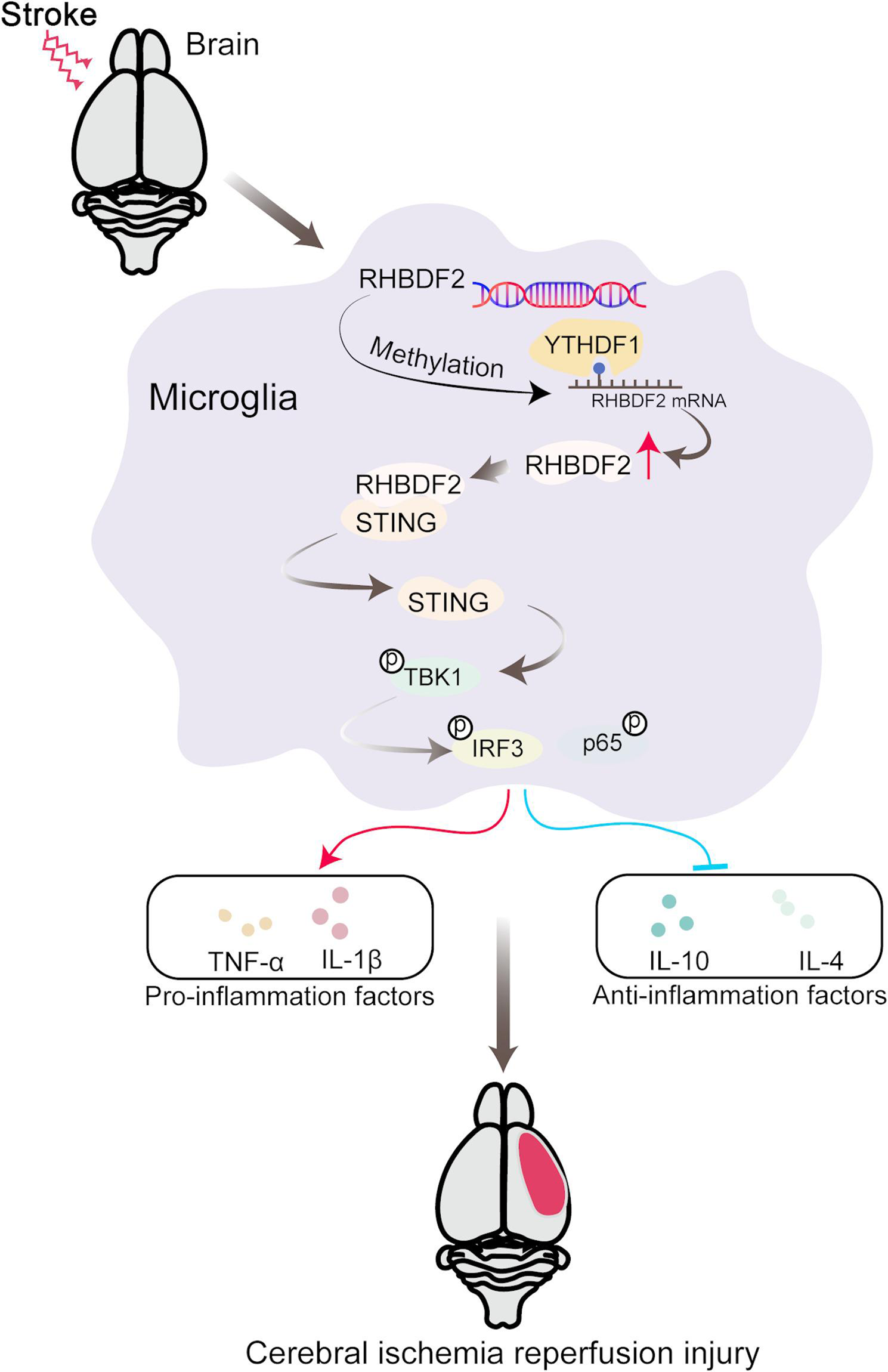
Adeno-associated viral (AAV) constructs carrying Iba1 promoter with shRHBDF2 (shRHBDF2AAV9) or shNC (shNCAAV9) sequence were packaged into recombinant AAV9 viral particles to achieve conditional knockdown of RHBDF2 in microglia for in vivo study. Groups of mice were given stereotaxic injections of either shRHBDF2AAV9 (2 µL, 1013 vg/mL) or shNCAAV9 into the cerebral cortex (from bregma: 0.25 mm posterior, 1.5 mm ventral, and 3 mm lateral). Downregulation of RHBDF2 expression in HMC3 cells was achieved with the recombinant lentiviral vectors (pLVX) carrying negative control (shNCLV) or shRHBDF2 (shRHBDF2LV) sequence.
Animal model and treatment
All in vivo experiments were performed in accordance with approved animal protocols and guidelines established by the General Hospital of Northern Theater Command (No.2023–26). A total of 128 mice were analyzed in this study. The middle cerebral artery occlusion/reperfusion (MCAO/R) mouse model was established with reference to a previous study (Bertrand et al. 2017). The nylon monofilament (0.21 mm in diameter, #45–0400) was used to induce focal cerebral ischemia. The nylon monofilament was placed in the middle cerebral artery. Mice experienced same surgery without vessel occlusion served as sham. After 90 min, the nylon monofilament was removed, and the mice were allowed to recover for 24 h. Three weeks before MCAO surgery, shNCAAV9 or shRHBDF2AAV9 viral particles (2 µL, 1 × 1013 vg/mL) were intracerebroventricular injected into mice (from bregma: 0.25 mm posterior, 1.5 mm ventral, and 3 mm lateral). Once the mice regained consciousness, their neurological function was assessed according to the study of Longa et al. (Longa et al. 1989). 0, no noticeable neurological deficits; 1, contralateral forelimb flexion; 2, circling toward the paralyzed side when walking; 3, leaning toward the side opposite the lesion; and 4, inability to walk spontaneously. A score between 1 and 3 indicated successful modeling.
2,3,5-triphenyltetraolium chloride (TTC) staining
After reperfusion, the brains were dissected and sliced to five thick coronal sections. The prepared 1% PBS-diluted TTC staining solution was added, and the section were incubated for 15 min in the dark at 37 °C. The sections were then photographed.
Fluro-Jade-C (FJC) staining
The degenerating neurons were detected with the FJC staining kit that purchased from Solarbio (#G3262, China). Briefly, the mouse brain tissue sections were prepared, dewaxed, and incubated with potassium permanganate solution for 10 min. FJC staining solution was prepared according to the manufacturer's instructions. After being incubated in FJC staining solution for 20 min, the sections were observed by fluorescence microscopy.
Cell culture
Human microglia cell line HMC3 cells were purchased from iCell (China) and maintained in MEM medium (Solarbio, China) supplemented with 10% fetal bovine serum (FBS, Tianhang, China). Cells were cultured at 37 °C in a humidified atmosphere.
The combination of oxygen and glucose deprivation (OGD) and reoxygenation was used to mimic CIRI in vitro (Sun et al. 2018). HMC3 cells were maintained in glucose-free medium (Minimum Essential Medium, Procell, China) and placed in a hypoxic environment for 4 h. The cells were then replaced with normal medium under standard culture conditions for 24 h.
For RHBDF2 knockdown, HMC3 cells were treated with shNCLV or shRHBDF2LV viral particles for 72 h, followed by OGD/R treatment. STING agonist, 1 μM diABZI (#GC35855, GLPBIO, USA), was added to the medium during OGD/R treatment.
Immunohistochemistry (IHC) staining
Paraffin-embedded brain sections were deparaffinized and dehydrated. Loss of antigenicity in sections was reversed with heated antigen retrieval solution for 10 min, and excess solution was blotted off. Peroxidase blocking was performed by incubating the sections for 15 min in 3% H2O2 solution. The sections were incubated with 1% bovine serum albumin (BSA, Sangon, China) for 15 min. The sections were incubated in primary antibody RHBDF2 (#AP13588A, Abcepta, China) overnight at 4 °C, and incubated with goat anti-rabbit IgG secondary antibody for 60 min at 37 °C. After being incubated with 100 μL DAB solution (#DAB-1031, MXB Biotechnology, China), the sections were counterstained with hematoxylin and photographed after mounting with neutral balsam.
Immunofluorescence (IF) staining
The prepared brain tissue sections were incubated with antigen retrieval solution and kept at high temperature for 10 min. The cell coverslips were fixed with paraformaldehyde and incubated with Triton X-100. The coverslips and sections were then blocked with 1% BSA for 15 min at room temperature. The STING (#19851-1-AP, Proteintech, China) primary antibody was then used to incubate the slides overnight. The slides were washed thrice with PBS for 5 min each. The Cy3- preabsorbed secondary goat anti-rabbit (IgG) diluted in PBS was then used to incubate the slides at room temperature for 60 min. After being counterstained with DAPI and applied one drop of anti-fade mounting medium, the slides were then observed under the microscope.
For double IF staining experiments, the sections were heated in an antigen retrieval solution and cooled to room temperature. After blocking endogenous peroxidase activity, the sections were incubated with 1% BSA for 15 min. The sections were incubated in the mixture of primary antibodies overnight at 4 °C. Then, the sections were then incubated with secondary antibodies for 90 min. After being counterstained with DAPI and applied one drop of anti-fade mounting medium, the slides were then examined using the microscope. The following combinations and concentrations of antibodies were used: Iba-1 (#ab283319, 1:200, Abcam, UK), CD16 (#16559-1-AP, 1:100, Proteintech, China), CD206 (#18704-1-AP, 1:100, Proteintech, China).
TUNEL-IF staining
Paraffin-embedded brain sections were deparaffinized and dehydrated. For permeabilization, the sections were incubated with 0.1% Triton X-100. After being incubated with antigen retrieval solution, the sections were incubated with TUNEL solution (Roche, Switzerland) for 60 min in the dark at 37 °C. The sections were then incubated with 1% BSA for 30 min in a humid environment. The primary antibody NeuN (#ab104224, Abcam, UK) was then used to incubate the slides overnight. The slides were then incubated with the secondary antibody for 60 min at room temperature. The slides were then observed under the microscope.
Immunoprecipitation and western blot analysis
For immunoprecipitation, the protein sample was isolated with Native Lysis Buffer (Solarbio) containing protease inhibitor cocktail (Solarbio). The supernatants were incubated with anti‐STING antibody or anti‐IgG antibody conjugated AminoLink Coupling Resin (Thermo) for 2 h. After being incubated, the bead-bound proteins were dissociated and further analyzed using western blot analysis.
The protein sample of ischemic mouse cerebral tissues and HMC3 cells was homogenized with RIPA buffer (Solarbio) containing protease inhibitor cocktail (Solarbio) and phosphatase inhibitor cocktail (Solarbio). After treatment with ice-cold lysis buffer for half an hour, the lysates were centrifuged at 10000 g for 5 min at 4 °C. For the extraction of total mitochondrial proteins from HMC3 cells, the mitochondrial protein extraction kit (Solarbio, #EX1110) was utilized. Protein samples were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF, Millipore, USA) membranes. After being blocked with blocking buffer (Solarbio), the membranes were incubated with primary antibodies diluted in antibody dilution buffer (Solarbio) at 4 °C overnight. The membranes were incubated with secondary antibodies for 1 h at 37 °C, and the membranes were developed using ECL western blotting substrate (Solarbio). Primary antibodies were used as follows: RHBDF2 primary antibody (Abcepta, #AP13588A), STING primary antibody (Proteintech, #19851-1-AP), YTHDF1 primary antibody (Proteintech, #17479-1-AP), TBK1 primary antibody (Affinity, #DF7026), p-TBK1Ser172 primary antibody (Affinity, #AF8190), NF-kB p65 primary antibody (Affinity, #AF5006), p-NF-kB p65Ser536 primary antibody (Affinity, #AF2006), IRF3 primary antibody (Affinity, #DF6895), phosphorylated IRF3 (p-IRF3Ser396) primary antibody (Affinity, #AF2436), Arg1 primary antibody (Affinity, #DF6657), iNOS primary antibody (Affinity, #AF0199), Cytochrome C primary antibody (Affinity, # AF0146), and COX4 primary antibody (GeneTex, #GTX49132).
Quantitative real-time PCR (qPCR)
RNA samples were extracted using TRIpure (BioTeke, China) and then reverse transcribed to cDNA using All-in-One First-Strand SuperMix (Magen Biotech, China). qPCR was performed using 2 × Taq PCR MasterMix and SYBR Green that were purchased from Solarbio. Primers for qPCR were as follows. Mus RHBDF2-F, AGCCCTCATCCTCGTGTC; Mus RHBDF2-R, CTGGTCTAGCTCGTACTTCTCA. Mus β-actin-F, GCCAGAGCAGTAATCTCCTTCT; Mus β-actin-R, AGTGTGACGTTGACATCCGTA. Homo RHBDF2-F, TGCCTCGTGTCTGTGGTCT; Homo RHBDF2-R, CGGTATGGGAGAAAGATGG. Homo YTHDF1-F, CAATGAGGCTCCGTGGTC; Homo YTHDF1-R, AAACAGCATCGTGCATAAAA. Homo β-actin-F, TCAGGGTGAGGATGCCTCTC; Homo β-actin-R, CTCGTCGTCGACAACGGCT.
Cytokines determination
The levels of cytokines in brain tissue homogenates and cell supernatants were measured using ELISA kit that purchased from Multi Sciences Biotech Co., Ltd (China) according to the manufacturer's instructions. The concentrations of pro-inflammatory and anti-inflammatory cytokines including TNF-α (#EK282 for mouse, #EK182 for human), IL-10 (#EK210 for mouse, #EK110 for human), IL-1β (#EK201B for mouse, #EK101B for human), IL-6 (#EK106 for mouse), and IL-4 (#EK104 for human) in mouse brain samples and cell supernatants were measured with the corresponding ELISA kits.
Flow cytometry
To detect the polarization of HMC3 cells, the cells were centrifuged (300 g for 5 min), washed, and resuspended. The resuspended cells (1 × 106) were incubated with 5 μL CD16-PE (F1101602, Liankebio, China) or 4 μL CD206-FITC (FITC-65155, Proteintech Group, USA) antibody at 4 °C in darkness for 30 min. The stained cells were washed, resuspended, and read on the NovoCyte flow cytometer (Aglient, USA). After adding DCFH-DA, fluorescence microscopy was used for imaging, and flow cytometry was used to quantify the fluorescence intensity of 2,7-dichlorodihydrofluorescein (DCF), a detector of ROS.
Methylated RNA immunoprecipitation (Me-RIP) PCR assay
The Me-RIP assay was performed using riboMeRIPTM m6A Transcriptome Profiling Kit according to manufacturer’s instructions. Total RNA (1 μg/μL) was fragmented into about 100 bp ~ 150 bp. For the preparation of Me-RIP beads, 25 μL magnetic beads A/G were incubated with 5 μg anti-m6A antibody in 250 μL IP buffer for 30 min at room temperature. IP reaction mixture (500 μL, containing fragmented RNA) was used to resuspend the prepared beads and incubated at 4 °C for 2 h. After being washed in IP buffer, the reaction mixture was eluted at 4 °C for 1 h with continuous shaking. The eluted RNA was further purified using the Magen Hipure Serum/plasma miRNA Kit. RNA m6A modification was then tested by qPCR assay. Primers for qPCR were as follows. Mus RHBDF2-5’UTR-F, CACAGCCCAACCGTCCA; Mus RHBDF2-5’UTR-R, AGGCAGAAGAGGCACCG. Mus RHBDF2-CDS-F, GGGCAAGCGACAAAACTG; Mus RHBDF2-CDS-R CCTGGAAGGATGGCACCT. Homo RHBDF2-CDS-F, AGCGAGACCTGGAGCGG; Homo RHBDF2-CDS-R, AGGGGCGGCCCTTGATC.
RNA immunoprecipitation coupled with PCR
YTHDF1 RIP was performed using the EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to the manufacturer’s protocol. For each reaction, 5 μg of anti-YTHDF1 antibody or the same amount of IgG was used.
ATP measurements
Relative ATP content in HMC3 cells was measured with the ATP assay kit (Beyotime, #S0026) according to the manufacturer's instructions. Briefly, cells were treated with lysis solution and incubated. After the incubation, cells were centrifuged at 12,000 g for 5 min at 4 °C, and the supernatant was reserved. ATP assay working solution prepared and stand at room temperature for 5 min. ATP assay working solution (100 μL) and the prepared samples (20 μL) were mixed and detected immediately after 2 s interval. The samples corresponding luminescence were measured in microplate reader at 570 nm.
RNA-sequencing
Total RNA from RHBDF2-knockdown HMC3 cells and its control cells were constructed and sequenced using the Illumina platform. The DEGs were generated using a cutoff value of|Log2Foldchange|> 1 and p. value < 0.05. The fragments per kilobase million (FPKM) values of each gene from different groups were used for gene set enrichment analysis (GSEA) by clusterProfiler. The protein–protein interaction (PPI) network of DEGs was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/). The top 10 hub genes were visualized using Cytoscape according to the identification of extracted PPI pairs. The relative abundance and proportion of immune cells in each sample was evaluated using CIBERSORT. Correlation between hub genes was generated using the corrplot package.
Proximity ligation assay
Proximity Ligation Assay (PLA) to visualize protein–protein interactions was performed using the Duolink® In Situ Detection Reagents Red DUOLINK (#DUO92008) according to the manufacturer’s protocol. Briefly, after OGD/R treatment, cells were fixed for 15 min with 4% PFA and washed 3 times with PBS. Cells were then permeabilized using 0.1% Triton X-100 for 30 min and then blocked for 15 min at room temperature using 1% BSA. Cells were then incubated overnight at 4 °C in STING antibody (Proteintech, #66,680–1-Ig) or RHBDF2 antibody (Thermo, # PA5-121,462). The next day, cells were incubated in anti-mouse plus (#DUO92002) and anti-rabbit minus (#DUO92004) proximity ligation assay probes diluted 1:5 in the provided antibody diluent. Cells were then washed, and then the ligase was added to cells for 30 min at 37 °C. This step was followed by further washing with PBS, prior to addition of the polymerase (diluted in amplification buffer, 1:80) for 100 min at 37 °C. Subsequently, cells were washed using PBS, then DAPI was used for nuclear staining. All samples were analyzed using a confocal microscope.
Statistical analyses
Dates are presented as mean ± SD. Two-tailed Student’s t-test between two groups. Experiments containing more than two groups were analyzed with one-way ANOVA, followed by Tukey's or Dunnett’s multiple comparison tests as requested. All these statistical analyses and graphic representations were obtained by using the GraphPad Prism software.




Comments (0)