RB photothrombotic rat model
Male Sprague–Dawley rats aged 7–8 weeks were housed under conditions with diurnal lighting and had access to food and tap water ad libitum. All animal procedures adhered strictly to the recommendations outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH, USA, 2013) and followed the ARRIVE guidelines (http://www.nc3rs.org/ARRIVE, accessed on August 31, 2021). The animal protocol underwent ethical review and was approved by the INHA University Institutional Animal Care and Use Committee (INHA-IACUC) prior to implementation (approval number INHA180105-531-2). Surgical procedures for the PTS animal model were performed according to Schmidt et al. (2012) with some modification. Briefly, 8-week-old male Sprague–Dawley rats (250–300 g) were anesthetized with an intramuscular injection of a ketamine (3.75 mg/100 g body weight) and xylazine hydrochloride (0.5 mg/100 g body weight) mixture. Tail veins were dilated with warm water at 37 °C, and then RB (3 mg/kg) was injected into the tail vein using a 26 G syringe. After allowing 5 min for RB to circulate to the brain, the rats were positioned in a stereotactic frame, and the scalp was longitudinally incised and retracted to expose the skull. Two laser beams, each with a diameter of 5 mm and a wavelength of 100 W, were stereotactically positioned onto the skull—one 0.5 mm anterior to the bregma and 3.5 mm lateral from the midline, and the other at a corresponding position on the opposite side. The skull was illuminated for 20 min, and the incision site was sutured. The control group received only an infusion of RB without light exposure. A laser Doppler flowmeter (Periflux System 5000; Perimed, Jarfalla, Sweden) was used to monitor regional cerebral blood flow (CBF) and relative CBF, and a thermoregulated heating pad and a heating lamp were used to maintain a rectal temperature of 37.0 ± 0.5 °C.
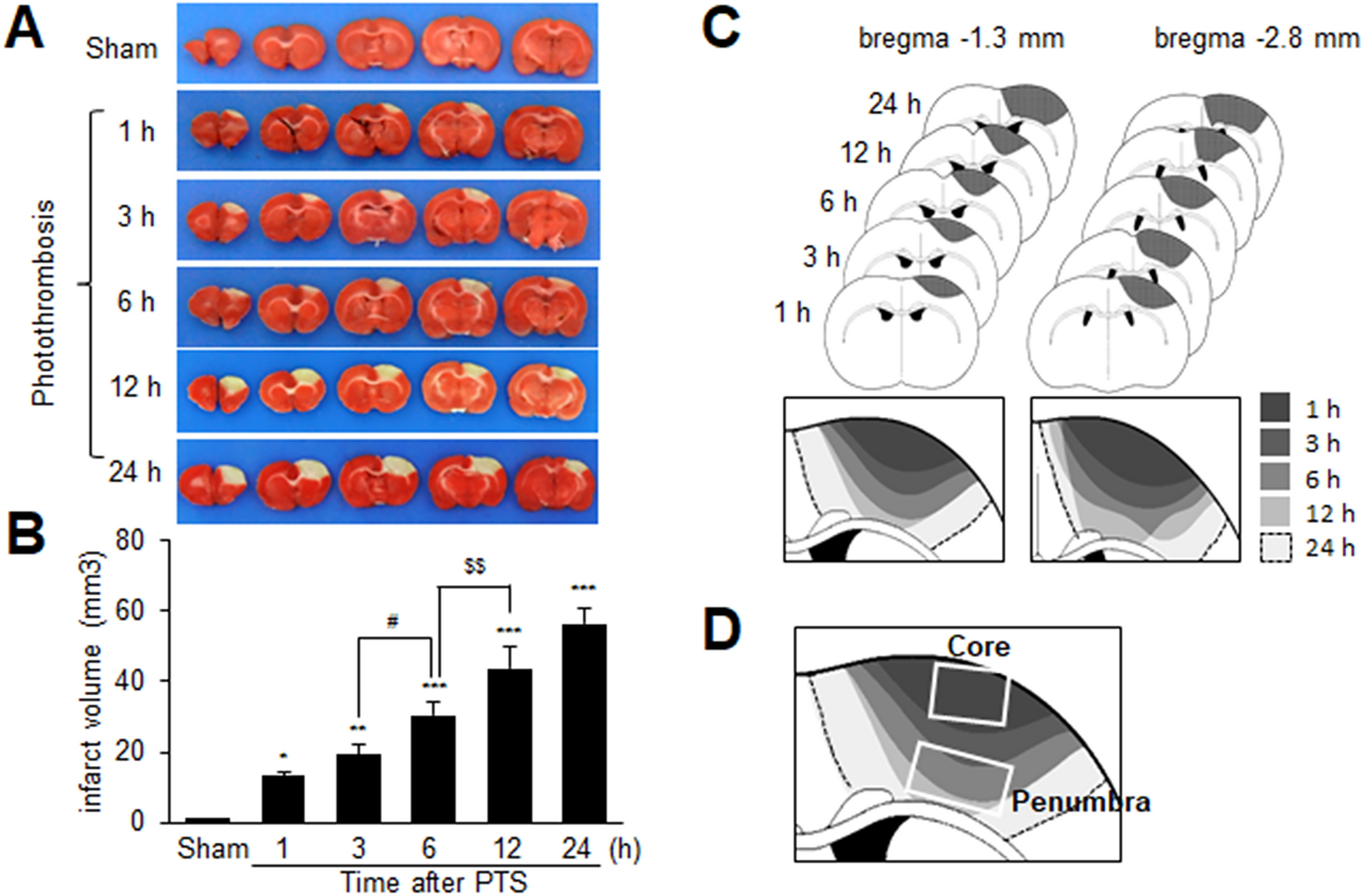
Infarct volume assessment
Rats were sacrificed, and their whole brains were dissected coronally into 2-mm slices using a metallic brain matrix (RBM-40000, ASI, Springville, UT, USA). The slices were immediately incubated in saline containing 2,3,5-triphenyl tetrazolium chloride (TTC 2%) at 37 °C for 15 min and then fixed in 4% paraformaldehyde (PFA, FUJIFILM Wako pure Chemical, Osaka, Japan). Scion Image (Scion Corporation, Frederick, MD, USA) was used to measure infarcted tissue areas. To adjust for edema and shrinkage, the areas of ischemic lesions were calculated using the formula: contralateral hemisphere volume—(ipsilateral hemisphere volume—measured injury volume). The infarct volumes were quantified (mm3) by summing the infarct sizes in adjacent tissue sections.
Modified Neurological Severity Score (mNSS)
Neurological deficits were assessed using the mNSS at 1-day post-PTS. The mNSS evaluates motor, sensory, balance, and reflex functions, with scores ranging from 0 to 18 (0 indicates normal function and 18 indicates maximal deficit) (Chen et al. 2001). Motor function was evaluated using two tests: (1) suspending the rat by its tail and scoring forelimb flexion, hindlimb flexion, and head movement > 10° with respect to the vertical axis on a scale from 0 to 1 (total score 0–3) within 30 s; and (2) placing the rat on the floor and scoring walking ability from 0 to 3 (0 for normal walking, 1 for inability to walk straight, 2 for circling toward the paretic side, and 3 for falling on the paretic side). Sensory function was tested using a placement test and a proprioceptive test, each scored from 0 to 1. Balance was assessed using the beam balance test, with scores ranging from 0 to 6 based on performance: 0 for maintaining a steady posture, 1 for grasping the side of the beam, 2 for hugging the beam with one limb off, 3 for hugging the beam with two limbs off or rotating around the beam for over 60 s, 4 for falling off the beam within 20–40 s, 5 for falling off within 20 s, and 6 for making no attempt to balance or hang onto the beam. Reflexes were scored on four items (maximum possible score of 4): pinna reflex (0–1); corneal reflex (0–1); startle reflex (0–1); and the presence of seizures, myoclonus, or myodystony (0–1).
Drug administration
Drugs were administered intranasally, as previously described (Kim et al. 2018). Rats were anesthetized via intramuscular injection of a mixture of ketamine (3.75 mg/100 g body weight) and xylazine hydrochloride (0.5 mg/100 g body weight). Following anesthesia, a nose drop containing HMGB1 A box (5 μg/kg; HM-012, HMGbiotech, Milano, Italy) dissolved in DW (100 µL) or BB-Cl-amidine (BBCA, 10 µg/kg; HY-111347A, MedchemExpress, NJ, USA) dissolved in DW (1 mL) was then carefully placed in each nostril of the anesthetized animals (positioned supine at a 90° angle) using a preautoclaved pipet tip (T-200-Y; Axygen, Union, CA, USA). The administration procedure was repeated until the entire dose was administered, with 2-min intervals between applications.
Serum and brain tissue sample preparation
The frontal cortices and striata of rat brains were isolated and frozen in liquid nitrogen for subsequent experiments after anesthetizing the mice with an intramuscular injection of a mixture containing ketamine (100 mg/kg body weight) and xylazine hydrochloride (23.32 mg/kg body weight). For serum samples, blood was collected through cardiac puncture using a 23 G syringe and left at room temperature for 30 min. Afterward, the blood samples were centrifuged at 6000×g and 4 °C for 10 min. The supernatant was then aliquoted and stored at − 80 °C.
Isolation of circulating neutrophils
Circulating neutrophils were isolated from the collected blood samples using gradient density centrifugation with Histopaque solution (Sigma-Aldrich, St. Louis, MO, USA), following a previously described protocol (Kim et al. 2019). Initially, Histopaque 1077 (3 mL) was layered on Histopaque 1119 (3 mL) in a 15 mL polypropylene tube and collected blood (3 mL) was carefully added on top of this mixture (Histopaque 1077/1119). The layered system, composed of three components, was centrifuged at 400×g for 30 min at room temperature. After centrifugation, the first ring of cells (mononuclear cells) was discarded, and the second ring (neutrophils) was transferred to a fresh 15 mL polypropylene tube containing phosphate-buffered solution (PBS) with 0.1% bovine serum albumin and 10% glucose (PBS-BG). The suspension was centrifuged at 1500 × g for 10 min at room temperature. To eliminate residual red blood cells, the resulting pellet was resuspended in 3 mL PBS-BG, poured onto Histopaque-1119 (3 mL), and centrifuged at 1500×g for 10 min at room temperature. The neutrophil ring was then transferred to a fresh 15 mL polypropylene tube containing PBS-BG and centrifuged at 1500×g for 10 min at room temperature. Finally, the pellet was resuspended in RPMI (Gibco BRL, Gaithersburg, MD, USA) containing 1% FBS.
Isolation of platelets from rat blood samples
After blood was drawn, it was placed into a Becton Dickinson Vacutainer® (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) containing Acid Cutrate Dextrose with a yellow cap. The samples were gently mixed after blood collection by slowly inverting the Vacutainer®. The blood was then centrifuged at 200×g for 20 min to obtain platelet-rich plasma (PRP), which was subsequently centrifuged at 100×g for 10 min in HEPES buffer [140 mM NaCl, 2.7 mM KCl, 3.8 mM HEPES, 5 mM EGTA (pH 7.4)] in the presence of 1 μM prostaglandin E1 (PGE1; Sigma-Aldrich, St. Louis, MO, USA). Platelets were pelleted by centrifuging the supernatant at 800×g for 10 min at room temperature. The platelets were then washed three times with platelet wash buffer [10 mM sodium citrate, 150 mM NaCl, 1 mM EDTA, 1% (w/v) dextrose (pH 7.4)] in the presence of 1 μM PGE1. Finally, the resulting pellet was resuspended in RPMI medium (Gibco BRL) containing 1% FBS.
Platelet-induced NET formation in co-culture assay
Neutrophils were isolated from freshly collected whole blood and seeded onto 6-well plates (SPL Life Sciences, Gyeonggi, South Korea) at a density of 1 × 106 cells/mL. Activated platelets, generated by PTS, were adjusted to 1 × 108 cells/mL in RPMI (Gibco BRL) containing 1% FBS and placed into the insert. Neutrophils and activated platelets were co-cultured at a 1:100 ratio to induce NETs for 2.5 h at 37 °C in 5% CO2/95% air. Adenosine triphosphate (ATP; Sigma-Aldrich, St. Louis, MO, USA) was used as a positive control. PMNs were pre-treated with HMGB1 A box (100 ng/mL; HM-012, HMGbiotech) or A438079 (20 µM, Tocris Bioscience, Bristol, Avon, UK) for 30 min and then co-cultured with platelets. For signaling pathway experiments, PMNs were pre-treated with TLR4 antagonist (TLR4-IN-C34, 20 µM, Sigma-Aldrich), C-X-C chemokine receptor type 4 (CXCR4) receptor antagonist (AMD3100, 20 µg/mL, Sigma-Aldrich), and RAGE antagonist (FPS-ZM1, 150 nM, Sigma-Aldrich) and then co-cultured with platelets.
Immunoblotting
Brain homogenates were extracted using RIPA buffer [50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% NP40, 0.25% sodium-deoxycholate, 0.5% Triton X-100, 10% glycerol, and Complete Mini Protease Inhibitor Cocktail tablet (Roche Diagnostics, Basel, Switzerland)]. Cell or tissue extracts were then loaded on 9–13% Sodium Dodecyl Sulphate–Polyacrylamide Gel Electrophoresis gels and immunoblotted using the following primary antibodies: anti-HMGB1 (1:2000, ab18256, Abcam, Cambridge, UK), anti-citH3 (1:1000, ab5103, Abcam), anti-GAPDH (1:10,000, 14C10, Cell Signaling Technology, Danvers, MA, USA), anti-MPO (1:1000, PA5-16672, Invitrogen, Waltham, MA, USA), anti-CD42b (1:1000, 12860-1-AP, Proteintech, IL, USA) and anti-CD62P (1:200, sc8419, Santacruz, CA, USA). The blots were detected using anti-rabbit HP conjugated or anti-mouse HP secondary antibodies (1:4000, Merck Millipore, Burlington, MA, USA) and a chemiluminescence kit (Merck Millipore).
Immunofluorescence staining
Brains were fixed in 4% PFA solution for 2 days at 4 °C, post-fixed in a 30% sucrose solution at 4 °C, sectioned at 40 μm using a vibratome, and immunologically stained. Sections were then blocked with 5% FBS, 5% horse serum, and 2% albumin in 0.1% Triton X-100 for 1 h at room temperature. Primary antibodies aganist mouse anti-laminin (NBP2-42392; Novus Biologicals, Centennial, CO, USA), rat anti-Ly6g (ab25024; Abcam, Cambridge, UK), rabbit anti-citH3 (ab5103, Abcam), rabbit anti-HMGB1 (ab18256, Abcam), mouse anti-HMGB1 (ab190377; Abcam, Cambridge, UK) and anti-CD42b (12860-1-AP; Proteintech) were diluted to 1:200. After incubation with primary antibodies, the brain sections were washed with PBS and incubated with rhodamine-labeled anti-rabbit IgG for anti-citH3, FITC-labeled anti-rat IgG for anti-Ly6g, FITC-labeled anti-rabbit IgG for anti-HMGB1, and FITC-labeled anti-mouse IgG for anti-HMGB1 in PBS for 1 h. All antibodies were diluted 1:200 and purchased from Merck Millipore Corporation. The sections were mounted on slides with VECTASHIELD Antifade Mounting Solution containing DAPI (Vector Laboratories) and examined under a Zeiss LSM 510 META microscope (Carl Zeiss Meditec AG, Jena, Germany).
Immunofluorescence staining of platelets
Isolated platelets were cytocentrifuged onto glass slides at 800 rpm for 5 min at room temperature using a Cytospin system (ThermoFisher Scientific, Waltham, MA, USA). The platelets were fixed with 4% PFA for 20 min at room temperature, followed by overnight incubation in 1% PFA at 4 °C for enhanced fixation. The cytospin slides were blocked with 1% normal goat serum for 1 h at room temperature to reduce nonspecific binding. Subsequently, the slides were incubated with an anti-HMGB1 antibody overnight at 4 °C. Images were acquired using a Zeiss LSM 510 META confocal microscope (Carl Zeiss Meditec AG).
Enzyme-linked immunosorbent assay (ELISA)
Serum HMGB1 levels were assessed using ELISA kits (Cusabio, Houston, TX, USA), according to the manufacturer's instructions. For serum samples, whole blood was collected at 1, 3, 6, 12 and 24 h after PTS, and allowed to clot by leaving it undisturbed for 30 min at room temperature. The clots were then removed by centrifuging the samples at 6000 rpm for 10 min in a cold microcentrifuge. The supernatants were immediately transferred to clean polypropylene tubes, and their concentrations were determined using ELISA kits.
Quantification of serum DNA
The serum DNA content was quantified using a Quant-iT PicoGreen double-stranded DNA (dsDNA) assay kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and fluorescence was measured using a Gen5 microplate reader (BioTek, Winooski, VT, USA) at an excitation/emission wavelength of 504/523 nm.
Statistical analysis
Statistical analysis was performed using PRISM software 5.0 (Graph Pad Software). ANOVA was initially used, followed by the Newman–Keuls test. The data are presented as means ± SEMs, with the p-values < 0.05 considered statistically significant.




Comments (0)