
Remember me

Multi-omics studies have revealed IEC defects in human IBD5, and revealed the presence of undifferentiated IECs with drastically reduced expression of chloride absorptive transporters (e.g., SLC26A3 and CAR4), and components of the mucous layer (e.g., MUC2 and CEACAM1), which play key roles in regulating intestinal homeostasis. Notably, down-regulation of both Slc26a3 and Car4 has been observed in C. rodentium-infected susceptible mice (such as FVB10) suggesting that this model could be used to study intestinal epithelial dysfunction. To that end and to understand the translatability of this model to human disease, we conducted a proteomics analysis in C. rodentium-infected FVB mice and assessed protein expression changes for key proteins that regulate intestinal homeostasis in the model and compared these results with our previously published IBD proteomics dataset20.
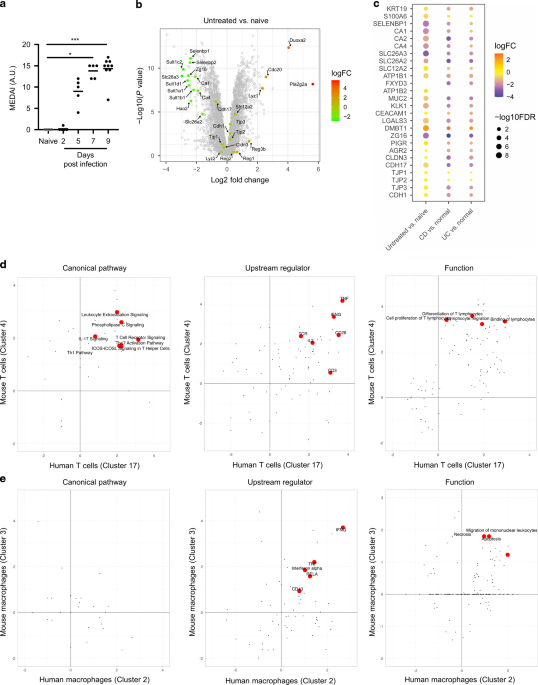
After inoculating FVB mice orally with C. rodentium, we confirmed an increase in fecal CFU on days 5 and 7 post-infection (p.i.) in colon tissue compared to non-infected control mice (Supplementary Fig. 1a). To better understand disease kinetics in infected mice, we established a novel endoscopy disease severity score, i.e., the Murine Endoscopic Disease Activity Index (MEDAI), which was defined based on thickening of the intestinal epithelium, presence of exudate and loss of vascularity (scoring system described in Supplementary Table 1). We observed an increase in the MEDAI as early as day 5 p.i., which peaked on day 7 p.i. and remained severe until day 9 p.i. (Fig. 1a; representative images shown in Supplementary Fig. 1b). Endoscopically, the colitis in C. rodentium-infected FVB mice was characterized by a white intestinal surface due to hyperplasia-induced increased granularity and release of exudate resulting in masking of vascularity (Supplementary Fig. 1b). By day 7 p.i, epithelial hyperproliferation led to angular protrusions of the thickened epithelium into the lumen resulting in a striated appearance, a hallmark of disease progression in this model (Supplementary Fig. 1b). Consistent with published literature21, we observed that 90% of the FVB mice exhibited severe clinical disease as manifested by mice that succumbed to infection or were euthanized after the loss of >20% body weight/moribund appearance by day 10 p.i. (Supplementary Fig. 1c). Disease kinetics reported by endoscopy were confirmed by histological analyses and Alcian blue staining of goblet cells, which showed progressive inflammation, mucosal hyperplasia and goblet cell loss in the distal colon (Supplementary Fig. 1d–1f). We observed that some crypts in CD and UC tissues, but not all, showed the loss of goblet cells, which is a sporadic feature that occurred in certain areas of the tissue (Supplementary Fig. 1g; indicated by the arrow), consistent with previous reports22,23,24.
Fig. 1: Factors critical for intestinal epithelial function exhibited similar protein expression changes and factors associated with T cell function, but not macrophage function, exhibited similar gene expression changes between C. rodentium-induced colitis and human IBD tissues.
a FVB mice were infected with C. rodentium p.o. and disease progression was measured using endoscopy at various times p.i. (n = 5–9 mice per time point). b Volcano plot depicting protein expression changes on day 8 p.i. compared to naïve mice detected using MS based proteomics. c Heat map of select protein expression changes in infected FVB mice with previously published CD or UC proteomics datasets20, compared to respective healthy controls. d IPA pathway, upstream regulator and function comparisons between mouse cell Cluster 4 and a human highly activated T cell cluster. e IPA pathway, upstream regulator and function comparisons between mouse cell Cluster 3 and a human inflammatory macrophage cluster. *p < 0.05, ***p < 0.001 determined as follows: Kruskal–Wallis with Dunnett’s multiple comparisons test (a). Data are representative of five independent experiments (a). Horizontal bars represent the mean of the points shown (a).
We performed a Mass-Spectrometry (MS) based global proteomics analysis of mouse colon tissues on day 8 p.i., at the peak of MEDAI, from naïve mice and untreated infected mice. As expected, a heat map showed that infection-induced major alterations in protein expression (Supplementary Fig. 2a). A volcano plot revealed that CAR4 and SLC26A3 were significantly downregulated compared with naïve controls (Fig. 1b), consistent with published data10. In diseased untreated mice, we observed a strong induction of the pro-proliferative/tumorigenic cycle regulator CDC20 and the Wnt signaling modulator PLA2G2A25. PLA2GA is expressed by secretory cells in the mouse colonic tissue and its secreted form, induced upon inflammation, drives proliferation of IECs in FVB mice25. Interestingly, PLA2G2A mRNA is increased in UC patient colonocytes compared to controls5. In our study, PLA2G2A protein level was not altered in human diseased tissues (data not shown). We also observed down-regulation of the SULT1 family of proteins and HAO2, which have been observed in other models of colitis, thus highlighting similarities between pre-clinical models of disease26.
Among the proteins critical for intestinal homeostasis, we analyzed changes in chloride absorptive transporters (SLC26A3 and CAR 1/2/4), a chloride secretory transporter (SLC12A2), components of the Na+/K+-ATPase (ATP1B1/B2) that establish electrochemical gradients critical for movement of Na+, K+ and Cl−, mucus components (MUC2, KLK1, CEACAM1, LGALS3, DMBT1, PIGR), a regulator of goblet cell differentiation (AGR2), differentiation markers for IECs (KRT19, S100A6, SELENBP1) and proteins involved in preventing bacterial invasion (DMBT1 and ZG16)5,27,28,29. Comparing mouse proteomics data to our published proteomics analyses in human IBD, we observed down-regulation of chloride absorptive machinery during disease in mice and humans, compared to healthy controls (Fig. 1c). In the same way, some components of the mucous layer i.e. MUC2, KLK1, LGALS3, DMBT1 and ZG16, also shared similar protein expression changes in infected mice and IBD tissues (Fig. 1c).
We examined expression of tight and adherens junction proteins that are parts of the IEC layer30. By proteomics, we found there was modest change in CLDN3, CDH1, CDH17 and TJP1-3 after infection (Fig. 1b) whereas they were more prominent changes in IBD patients (Fig. 1c). We also analyzed changes in proteins in involved wound repair31 and found that the extent of the protein changes was modest in both the mouse disease and human IBD (Supplementary Fig. 2b and Fig. 1c). While differences in fold expression between mouse and human diseased tissues as well as between UC and CD tissues were observed, we found that our proteomics analyses identified similar patterns of dysregulation of key intestinal homeostatic proteins involved in solute transport and mucous layer in C. rodentium-induced colitis in FVB mice and in human IBD.
We also assessed changes in disease relevant immune cells such as T cells and macrophages at the same time point when the IECs were assessed, to address whether they undergo similar changes in C. rodentium-induced colitis as human IBD. We performed cell type label transfer from CD patients32 to mouse cell clusters based on transcriptomic signatures and found that a majority of Cluster 4 cells are highly activated T cells in CD patients and that Cluster 3 cells were inflammatory macrophages in CD (Supplementary Tables 2 and 3). We then compared gene expression changes after C. rodentium infection in mouse T cells (Cluster 4) and macrophages (Cluster 3) to their counterpart cell populations in CD. IPA analysis revealed that several pathways were regulated similarly between mouse T cells after C. rodentium infection and human highly activated T cells from IBD patients (Fig. 1d). Such pathways included leukocyte extravasation, T cell receptor activation, and Th1 and Th17 responses (Fig. 1d). Mouse and human T cell changes shared similar upstream regulators and cellular functions such as cell migration, differentiation, and proliferation (Fig. 1d). In contrast to T cells, mouse and human macrophages had less similarities based on IPA pathway analyses, although mouse and human macrophages performed similar cellular functions as evidenced by shared function terms including cell migration (Fig. 1e). Future studies will address the alterations in immune cells that occur during the early phases and the impact of IL-22 treatment.
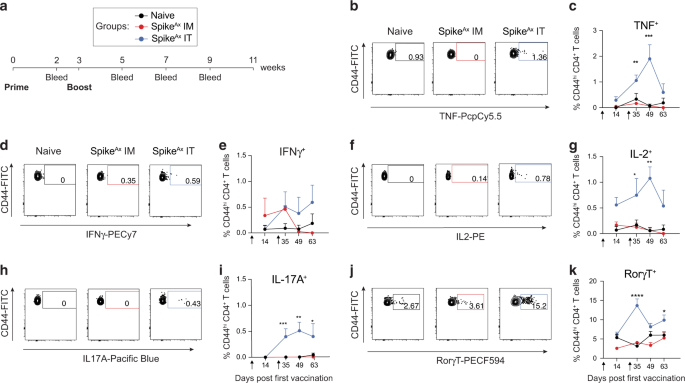
IL-22.Fc prevented colitis and improved survival in C. rodentium-infected FVB miceSeveral studies indicated that fatal colitis in FVB mice is due to infection-induced epithelial hyperplasia potentially due to an increase in Wnt signaling modulators, including RSPO2 and/or secreted PLA2G2A11,25. IL-22 inhibits Wnt and NOTCH signaling in mouse intestinal organoid cultures12. Moreover, due to the multi-faceted role of IL-22 in regulating several other aspects of intestinal epithelium homeostasis (reviewed in4,18), we assessed the secretion of IL-22 from colonic explants following C. rodentium-infected mice. We found that IL-22 secretion peaked on day 3 p.i. and decreased to basal level on day 7 and 9 p.i. from C. rodentium-infected mice when compared to naïve mice (Fig. 2a), consistent with a previous study33. The reduced levels of IL-22 C. rodentium-infected FVB on day 7 and 9 p.i. could be the cause of disease worsening in the infected FVB mice. This prompted us to determine whether exogenous IL-22.Fc would ameliorate disease in infected FVB mice and rescue them from lethality.
Fig. 2: Exogenous IL-22 treatment protected C. rodentium infected FVB mice from lethality.
Prophylactic treatment of IL-22.Fc was started before infection and administered twice per week until the end of the experiment (n = 3–10 mice per group). a IL-22 secretion from colonic explants of naïve non-infected mice and C. rodentium infected FVB mice at various times p.i. b Schematic of prophylactic treatment of IL-22.Fc. On day 5 p.i., fecal burdens (c), and on day 7 p.i. endoscopic disease severity (d), distal colonic inflammation (left) and hyperplasia (right) in H&E stained tissues (e) were measured. f Representative endoscopy (top) and H&E staining (bottom) images of distal colon in various treatment groups. Scale bar, 50 μm. g Percent survival curves from the start of the experiment until 90% of the untreated animals were either found dead or exhibited >20% body weight lost. h–j Immunoblot analyses of REG3B, REG3G, PHGR1 and GAPDH (loading control) in various treatment groups (data from individual mice shown in h and i) and densitometric quantification (j). NS, not statistically significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 determined as follows: One sample Wilcoxon test or Mann–Whitney test multiplied by the number of inter-group comparisons (c–e), log-rank test (g), or one-way ANOVA with Dunnett’s multiple comparisons test (j). Data are representative of two (a) or four independent experiments (c, d, f and g). Horizontal bars represent the mean of the points shown (c, d, e and j).
We started treatment with IL-22.Fc or vehicle control (PBS) intraperitoneally one day before C. rodentium infection and maintained treatment twice a week on days 2, 6, 9 and 13 p.i. (Fig. 2b). Exogenous IL-22.Fc reduced bacterial burden in the feces (Fig. 2c) on day 5 p.i. and reduced the endoscopic MEDAI compared with vehicle control on day 7 p.i. (Fig. 2d and f). Endoscopic measures of exudate, granularity and vascularity were all improved in IL-22.Fc-treated mice (Fig. 2d and f). Histologic evaluation confirmed a significant reduction in mucosal inflammation and hyperplasia in the distal and mid colon (Fig. 2e and f). IL-22 treatment also prevented the goblet cell loss, albeit with variability in the degree of amelioration (Supplementary Fig. 2c). IL-22.Fc treatment significantly enhanced survival in C. rodentium-infected FVB mice (Fig. 2g). Untreated and PBS treated mice lost significant more body weight than naïve animals on day 9 p.i., demonstrating the severity of disease in the infected animals (Supplementary Fig. 2d). Although the average body weights of IL-22 treated group were higher than those of untreated or vehicle control groups, the differences were not statistically significant consistent with other reports where IL-22 treatment had no impact on body weight34,35; however, this is in contrast to studies where exogenous IL-22 did improve body weight indicating that effect of IL-22 on body weight is context dependent36,37. Co-housing the IL-22 treated mice with vehicle treated mice conferred partial protection on vehicle treated mice (Supplementary Fig. 2e) consistent with the role of IL-22 in altering the endogenous microbiota to favor protection from pathogenic bacterial species15.
IL-22 drives host defense by eliciting the production of AMPs such as REG3B and REG3G in response to microbial infection4. Consistent with these published observations, we found that REG3B and REG3G protein levels were increased in IL-22.Fc-treated animals compared to vehicle control treated animals (Fig. 2h and j). Finally, to phenotypically assess the differentiation of intestinal epithelium, we evaluated expression of PHGR1 protein, which shows highest expression in the most differentiated cells of the human intestinal epithelium by IHC38. We found that PHGR1 was decreased in whole colonic tissue in infected untreated mice and IL-22.Fc-treatment restored the expression of PHGR1 to levels comparable to naïve mice (Fig. 2i and j). As such, IL-22 treatment potentiated host-defense, reduced disease severity and rescued FVB mice from C. rodentium infection induced lethality.
CITE-seq analysis revealed the emergence of undifferentiated intermediate IECs during disease, resolved the MoA of IL-22 in vivo and identified parallels between C. rodentium-induced colitis and IBDOvert epithelial hyperplasia in FVB mice during C. rodentium infection likely interferes with normal epithelial differentiation, resulting in undifferentiated IECs akin to those reported in human samples5. Moreover, how IL-22 treatment regulates the function of various IEC lineages in vivo is not well understood. Thus, using single-cell CITE-seq technology, we determined whether there was heterogeneity in IECs in various treatment groups, and determined how IL-22 treatment altered their transcriptomic signature.
Colon cells were isolated on day 12 p.i. and processed for CITE-seq. We identified and visualized 24 cell clusters by Uniform Manifold Approximation and Projection (UMAP) (Fig. 3a). We first performed automatic cluster annotation using SingleR39 by comparing transcriptomic similarities of single cells from individual clusters to reference databases. Based on the SingleR analysis (shown in Supplementary Table 2), we identified 8 different cell types distributed across various clusters: 1) epithelial cells, 2) macrophages/monocytes, 3) dendritic cells, 4) T/NKT/Innate Lymphoid Cells, 5) B cells, 6) fibroblasts, 7) endothelial cells, and 8) stromal cells. For further analyses we focused on epithelial cell clusters (Clusters 0, 12, 16, 22 and 23), which expressed higher levels of EPCAM protein detected using a CITE-seq specific barcoded antibody (Fig. 3a, inset). Based on similarities in the transcriptomic signature, we annotated mouse IEC clusters by transferring cell type labels from a UC study to mouse IEC Clusters 0, 12, 16 and 22, but not Cluster 23 due to the smaller number of cells compared to other clusters, which precluded statistical analysis (Fig. 3a; Supplementary Table 4)5. We found that the majority of cells in Clusters 0, 12, 16 and 22 were undifferentiated cells, goblet cells, colonocytes and enteroendocrine cells (EECs), respectively (Fig. 3a; Supplementary Table 4). All the clusters shared similar cell type markers as their counterparts in human tissues5 (Supplementary Fig. 3a), thus confirming our cell type label transfer.
Fig. 3: Heterogeneity of intestinal epithelial cells in C. rodentium induced colitis.
CITE-seq was conducted on lamina propria cells isolated from various treatment groups – infected (untreated, vehicle or IL-22.Fc treated) or naïve mice (n = 5 mice per group). a Integrated UMAP clustering of all different treatments highlighting the epithelial cell clusters that can be distinguished in the feature plot by expression of EPCAM antibody derived tag (inset). Clusters 0, 12, 16, 22 and 23: epithelial cells; Clusters 1, 3 and 9: macrophages/monocytes; Clusters 2, 8, 11 and 19: dendritic cells; Cluster 4: T cells, NKT cells and innate lymphoid cells; Clusters 5, 6 and 14: B cells; Clusters 7 and 10: fibroblasts; Clusters 15 and 21: endothelial cells; Cluster 17: stromal cells; Clusters 13, 18 and 20 contained mixed populations. b Pseudotime trajectory analysis of the IECs with stem cells (white), intermediate cells (Branched [B] or transitional [T]; grey), colonocytes (green), and goblet cells/EECs (blue). c Distribution of IEC types identified using pseudotime trajectory analysis in the various treatment groups.
Next, we performed pseudotime trajectory analyses to understand changes in IEC differentiation states in various treatment groups. In this analysis, we were unable to distinguish between goblet cells and EECs in Cluster 22 because of its small size, heterogeneous composition and due to overlapping secretory mechanisms between goblet cells and EECs, consistent with observations in human studies5. In all groups, trajectory analyses revealed a dichotomous trajectory of IECs developing from stem cells at Milestone S into two distinct differentiation lineages, i.e., absorptive, and secretory cells ending at Milestones C and G, respectively (Fig. 3b), as reported previously5. To elucidate the differentiation state of various clusters, we investigated their distribution within the trajectory. The majority of Cluster 16 was present at colonocyte Milestone C whereas Cluster 12 and Cluster 22 were at goblet cells/EECs Milestone G (Fig. 3b, c; Supplementary Table 5). Notably, after C. rodentium infection, intermediate states (Branching [B]/Transitional [T]) were present in both the colonocyte as well as goblet/EEC clusters in untreated or vehicle treated mice, indicative of infection-induced incomplete differentiation from progenitor cells (Fig. 3c; Supplementary Table 5). IL-22.Fc treatment restored the distribution of cells in the colonocyte cluster to a pre-disease state suggesting that Cluster 16 was highly responsive to IL-22. Using Cluster 12/22 and 16 as references, we studied the distribution of cell types within Cluster 0 and found that it contained mostly undifferentiated cells (i.e., stem cells or intermediate cells), some colonocytes but not goblet cells/EECs (Fig. 3b, c; Supplementary Table 5). Finally, to appreciate the degree of alteration in the overall landscape of IEC differentiation during each treatment condition, we combined the data from IEC clusters. After infection, there was an increase in the percentage of intermediate cells from 37.0% in naïve FVB mice to 51.2% in C. rodentium-infected FVB mice (Fig. 3c). IL-22.Fc treatment decreased the prevalence of intermediate cells to 28.3% compared with vehicle controls at 47.0% (Fig. 3c). To further understand the nature of intermediate IECs, we isolated Cluster 0 cells from infected untreated FVB mice in silico, since majority of the intermediate cells were undifferentiated Cluster 0 cells (Fig. 3c; Supplementary Table 5). We identified four subclusters in total (Supplementary Fig. 3b). The UC study reports five sub-cell types from human undifferentiated IECs including stem cells, early transit-amplifying cells, cell-cycle cluster cells (transit-amplifying-like-cells with high expression of cell cycle genes), absorptive progenitor cells and secretory progenitor cells5. We, therefore, annotated subclusters of the mouse undifferentiated cells by transferring sub-cell type labels from the UC study to mouse cells. Interestingly, we found all the four mouse subclusters were mixed cell populations of early transit-amplifying cells and absorptive progenitor cells (Supplementary Table 6).
In summary, we identified mouse IEC clusters that were akin to those observed in human samples5, thus underscoring the translatability of this model to human disease. Infection increased the frequency of intermediate IECs that have not completed their differentiation into terminal lineages critical for intestinal epithelial function, which likely contributed to disease development. IL-22.Fc treatment prevented the accumulation of intermediate IECs that occurred during disease.
CITE-seq analysis of C. rodentium-induced colitis following IL-22 treatment revealed disease-relevant changes in gene expression of specific epithelial cell populations that were similar to pathogenic mechanisms in IBD patientsNext, we asked whether specific IEC clusters responded differently to IL-22.Fc treatment versus vehicle in diseased mice. Cluster 22 emerged in C. rodentium-infected FVB mice, albeit with very low numbers (Fig. 3c), and therefore, we focused on IEC Clusters 0, 12, and 16 for analyses of differentially expressed genes (DEGs) comparing IL-22 treated mice to vehicle (Fig. 4a–d) and untreated infected mice to naïve (Supplementary Fig. 4a–d). When compared to naïve mice, a heat map of the overlapping DEGs revealed that most, but not all, of the disease associated changes were reversed by IL-22.Fc treatment (Supplementary Fig. 3c). Among the top 50 DEGs, we identified several markers of terminally differentiated IECs as well as solute transporter genes (Supplementary Table 7). Some solute transporter changes overlap with those found in whole tissue microarray studies reported previously10, thus validating our transcriptomics analyses. Next, we assessed the differential expression of genes involved in intestinal homeostasis, some of which were highlighted in Fig. 1c.
Fig. 4: Genes modulated by IL-22 treatment at the single IEC level.
Heat maps of differentially expressed genes in IL-22.Fc treated mice compared to vehicle in various IEC clusters: IEC differentiation markers (a), AMPs (b), mucous layer genes (c), solute transporters (d). Clusters 0, 12 and 16 were undifferentiated cells, goblet cells and colonocytes, respectively.
Analyses of the differentiation markers reported in human IECs5 revealed that several genes had low to undetectable expression in mouse cells (Supplementary Fig. 3a). Among the detectable genes, we found that in infected untreated mice there was a decrease in markers that were highly expressed on differentiated human IECs, including Slc26a3, Car1, and Phgr1 in Clusters 0 and 16 when compared to naïve mice (Supplementary Fig. 4a). Of note, Phgr1 was among the top down-regulated genes in the undifferentiated and colonocyte cluster in diseased mice (Supplementary Table 7) validating the protein changes shown in Fig. 2i and j. IL-22.Fc treatment increased the expression of the majority of these differentiation markers including Slc26a3, Car1, Car4 and Phgr1 compared to vehicle (Fig. 4a). We did not observe a significant change in Alpi (Alkaline phosphatase, intestinal) either following the infection or after IL-22 treatment.
Among the AMPs, IL-22.Fc increased expression of Reg3b and Reg3g mRNA in all three IEC clusters (Fig. 4b), whereas Defb4540 and Rnase441 were only upregulated in undifferentiated cells. Strikingly, Wfdc2, a novel AMP reduced in UC patients5, was upregulated by IL-22.Fc in undifferentiated cells but was down-regulated in Cluster 16 colonocytes (Fig. 4b), thus highlighting cell type specific regulation of function. Since Cluster 0 is the largest IEC cluster (Supplementary Table 2), we speculate that there should be an increase in Wfdc2 expression in IL-22.Fc-treated animals.
For goblet cell differentiation and mucus layer integrity, we reviewed the genes involved in differentiation (Nupr1, Agr2, Klf4 and Elf3) or formation of critical components of the colonic mucus layer (Muc2, Muc3, Klk1, Oit1, Ceacam1, Mep1a, Lgals3, Dmbt1, Pigr and Pchd17)28,42,43,44,45,46,47,48. Except for Muc2, all the other genes were significantly up-regulated with IL-22.Fc treatment in undifferentiated stem cells (Fig. 4c). IL-22.Fc treatment significantly increased Muc2, Nupr1 and Agr2 in goblet cells suggesting that IL-22 drives differentiation and function of goblet cells. In contrast, Agr2 expression was decreased in colonocytes, consistent with its role in regulating goblet cell function44,
Comments (0)