Recent research has increasingly highlighted the cardioprotective roles of Sig-1R in mitigating hypertrophy, cellular toxicity, apoptosis, and the pressure overload-induced maladaptive endoplasmic reticulum (ER) stress response [12]. Ischemic cardiac injury leads to hypoxia, resulting in a decline in oxidative phosphorylation, reduced cellular adenosine triphosphate (ATP) levels, and the disruption of mitochondrial membrane polarisation. Previous studies have demonstrated that Sig-1R activation mitigated oxidative stress [13, 14], suppressed intracellular Ca2+ overload [15], and activated the PI3K/Akt/eNOS pathway [16], potentially protecting against myocardial ischemia-induced injury. Although not many studies have discussed that Sig-1R stimulation can protect the injured myocardium post-ischemia–reperfusion by reducing apoptosis and improving endothelial integrity, Sig-1R agonists were administered before ischemic events in all these studies, potentially confounding the distinction between preventive and therapeutic effects and deviating from clinical reality, thereby reducing the translational relevance of such findings. Moreover, the lack of post-ischemic administration in these studies leaves a significant gap in understanding the true therapeutic potential of Sig-1R activation in a clinical setting, where intervention typically occurs after ischemia has started [17, 18]. To address this limitation and more closely mimic the clinical reality, we started administering fluvoxamine, a well-established Sig-1R agonist, one day after the induction of myocardial ischemia–reperfusion in rats to simulate ischemia and drug administration early after ischemia injury in a realistic context. Our results demonstrated that Sig-1R stimulation by fluvoxamine contributed to LV remodelling suppression and LVEF recovery with myocardium salvage post-ischemia–reperfusion. Furthermore, these cardioprotective effects were sustained even after fluvoxamine was discontinued.
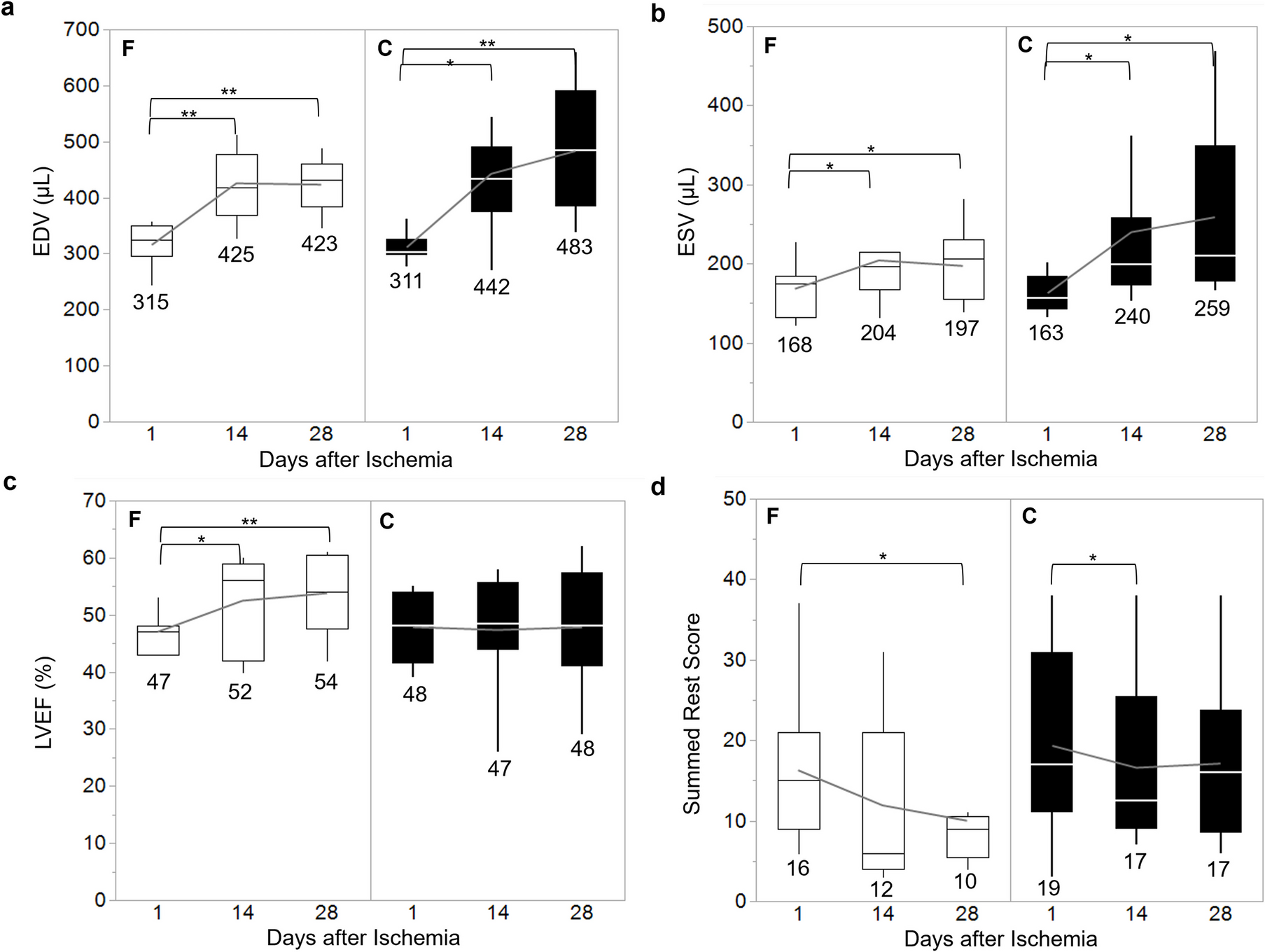
It was observed that in the fluvoxamine-treated group, Sig-1R stimulation resulted in a significantly greater proportion of salvaged myocardium compared with the control group, despite both groups exhibiting similar AAR percentages. The SRS, derived from myocardial perfusion imaging using 99mTc-MIBI ECG-gated SPECT, which quantifies the extent of myocardial perfusion defects at rest, showed improvement in both groups two weeks post-injury, suggesting that the heart’s inherent self-repair mechanisms were at play. However, in the fluvoxamine-treated group, the protective effects persisted for up to two weeks after treatment discontinuation, indicating a sustained therapeutic benefit in mitigating post-ischemia myocardial damage.
Our previous research has established 125I-OI5V as a valuable agent for visualising cardiac Sig-1R expression. It has been demonstrated that Sig-1R accumulation occurs particularly in the severely damaged myocardium surrounding non-salvaged areas, peaking around the third day following ischemia–reperfusion and gradually declining by 28 days [9, 11]. Building upon these findings, the current study further highlights the temporal dynamics of Sig-1R activation in mediating cardioprotective effects. Specifically, at 28 days post-ischemia–reperfusion, we observed that the 125I-OI5V uptake ratio of the AAR to the normally perfused area correlates positively with the 201TI uptake ratio of the AAR to the normally perfused area and the area ratio of the salvaged area to the whole LV in the fluvoxamine group. In contrast, no such correlations were present in the control group. These findings, when considered alongside prior observations of an inverse linear relationship between the 125I-OI5V uptake ratio and the 201Tl uptake ratio observed three days post-ischemia–reperfusion without intervention [11], suggested that Sig-1R expression initially increased as a compensatory response to ischemia injury, particularly in severely damaged regions. However, without external intervention, this response was insufficient for long-term and sustained cardioprotection, resulting in progressive cell death. By contrast, fluvoxamine administration early after ischemia injury extended the cardioprotective effects of Sig-1R activation, ultimately leading to greater myocardial salvage. Interestingly, many studies that utilised Sig-1R stimulators have reported a significant upregulation of Sigma-1 receptor expression [6,7,8, 17, 19]. However, in our study, no significant difference in Sig-1R expression was observed. This discrepancy may be attributed to the difference in treatment duration. Unlike studies with continuous drug administration over 28 days, the fluvoxamine treatment in our study was halted 14 days post-ischemia–reperfusion. The natural decline in Sig-1R stimulation post-treatment likely diminished fluvoxamine’s impact, resulting in comparable Sig-1R levels at the 28-day post-ischemia and reperfusion time point.
Initially, M1 proinflammatory macrophages dominate, exacerbating tissue damage through their release of inflammatory cytokines. This phase is followed by the recruitment of M2 anti-inflammatory macrophages, which facilitate tissue repair and contribute to collagen deposition [20, 21]. While the relationship between Sig-1R and macrophage infiltration post-ischemia–reperfusion is not yet fully understood, several studies suggest that Sig-1R plays a key role in neuron-macrophage interactions. For example, the absence of Sig-1R has been shown to reduce macrophage and monocyte infiltration after spared nerve injury, indicating that Sig-1R may influence immune cell recruitment [22, 23]. However, in our study, macrophage infiltration in the myocardium significantly decreased following the administration of fluvoxamine, which contrasts with the findings in the nervous system. These differences could be explained by the tissue-specific role of Sig-1R. In the nervous system, Sig-1R deficiency may impair neuron-macrophage communication by reducing the levels of C–C motif chemokine ligand 2 (CCL2), a key chemokine that mediates macrophage and monocyte recruitment by injured neurons in the dorsal root ganglion (DRG), leading to reduced infiltration of these immune cells [22]. Meanwhile, in the heart, Sig-1R activation likely attenuates inflammation by restoring the activity of protein kinase B (Akt) and endothelial nitric oxide synthase (eNOS) in cardiomyocytes, effectively suppressing macrophage infiltration and polarisation, thereby minimising further tissue damage [24,25,26]. Our previous study [11] demonstrated that three days post-ischemia, Sig-1R expression closely corresponded to macrophage infiltration. This pattern was corroborated in the present study, where regions with high CD68 expression primarily contained areas of elevated 125I-OI5V uptake. This consistent colocalisation across the early and late stages post-ischemia suggests a potential association between Sigma-1 receptor expression and the microenvironment supporting macrophage activation or recruitment. Such distribution may indicate a role for Sig-1R in modulating local inflammation or facilitating tissue repair in the affected areas.
Sig-1Rs are predominantly localized at the MAM, a critical subcellular domain involved in calcium signaling, mitochondrial homeostasis, and redox regulation. This spatial positioning enables Sig-1R activation to mitigate oxidative stress and preserve mitochondrial integrity. In models of pressure overload, Sig-1R activation has been shown to attenuate cardiac hypertrophy and improve contractile function through the Akt-eNOS signaling pathway [7, 27, 28]. Given the overlap in downstream pathological mechanisms, it is conceivable that the cardioprotective effects of fluvoxamine observed in the present study might, at least in part, be mediated through reactivation of this pathway.
Furthermore, evidence from the nervous system indicates that Sig-1R activation enhances Nrf2 signaling and suppresses NF-κB activity [29,30,31], thereby modulating oxidative and inflammatory responses. Supporting this, independent studies in sepsis models have demonstrated that Sig-1R activation reduces mitochondrial oxidative stress and myocardial injury via the Nrf2/HO-1 signaling axis [32]. Collectively, these findings reinforce the role of Sig-1R as a key regulator of redox homeostasis and cellular protection under pathological stress. Future investigations should aim to elucidate whether these pathways similarly contribute to cardioprotection following ischemia–reperfusion and to explore the therapeutic potential of targeting Sig-1R in ischemic heart disease.
Cardiac fibrosis is a critical pathological repair process observed in various cardiovascular diseases, contributing to heart failure progression, structural deterioration, and, in extreme cases, cardiac rupture [33]. Qu et al. demonstrated the protective efficacy of Sig-1R in cardiac fibrosis using a transverse aortic constriction (TAC) model, showing that Sig-1R offers protection by modulating the activation of cardiac fibroblasts, primarily through IRE1 pathway inhibition and autophagic flux restoration [8]. Consistent with these findings, our study revealed that Sig-1R stimulation significantly reduced the extent of cardiac fibrosis compared with the non-intervention group, highlighting the antifibrotic effects of Sig-1R post-ischemia–reperfusion.
In addition to structural changes, this study demonstrated an improvement in cardiac function. With the administration of fluvoxamine, although the EDV and ESV continued to increase post-ischemia–reperfusion, the rate of this increase was significantly decreased, resulting in a significant improvement in LVEF. In contrast, the control group showed minimal changes in LVEF. Remarkably, this trend persisted for up to two weeks after treatment discontinuation. Similar findings have been reported in other studies conducted using pure MI models with full-course medication, where fluvoxamine administration improved the maximal rate of pressure rise and decline (dP/dtmax and dP/dtmin) and enhanced fractional LV shortening [6, 19].
These findings suggest that fluvoxamine plays a pivotal role in mitigating myocardial dysfunction and preventing adverse remodelling through enhanced myocardial salvage post-ischemia–reperfusion. Furthermore, the prolonged therapeutic effects observed even after treatment cessation highlight its potential for promoting sustained cardiac recovery. These results position fluvoxamine as a promising therapeutic candidate for ischemic heart disease, warranting further investigation to fully elucidate its long-term benefits and underlying mechanisms.
This study has several limitations that are worth mentioning. First, the relatively small sample size may have reduced the statistical power to detect subtle differences between groups; therefore, studies with larger cohorts should be conducted in the future to address this limitation. The reduced sample size was primarily due to mid-experiment mortality and the exclusion of data that showed excessive variability, which could introduce bias. Consequently, more robust experiments with improved consistency will be necessary to investigate the trends observed in this study further. Second, fluvoxamine was administered for only two weeks during the four-week observation period. While this timeframe was chosen based on the hypothesis that short-term interventions could induce sufficient cardioprotective effects, a longer treatment duration might provide additional insights into fluvoxamine’s full therapeutic potential, particularly regarding its long-term effects on cardiac remodelling. The four-week observation period was designed to evaluate infarct healing, typically completed within this period, as well as to capture both the structural and functional changes in the myocardium after treatment cessation. However, a longer observation period could be considered in future studies to better assess the sustained efficacy of fluvoxamine in preventing adverse cardiac remodelling and improving long-term outcomes.




Comments (0)