Construct design and expression vectors
The N-terminal extension of bullfrog (Rana catesbeiana) ferritin lower subunit (res. 2–9, PDB ID: 1RCC [25]) with a point mutation N8Q to eliminate a potential N-glycosylation site was fused to Helicobacter pylori ferritin (res. 3-167, PDB ID: 3BVE [26]) with point mutations I7E to preserve the conserved salt bridge with bullfrog ferritin at residue 6R, and N19Q to remove a potential N-glycosylation site; this hybrid ferritin was previously designed by Kanekiyo et al. [11] to have antigen-presentation sites evenly distributed across its surface.
The Gn head domain sequence (res. 154–467, UniProt ID: P03518 [27]) was selected with point mutations E276G, L329H and I444V [28] and fused upstream to the hybrid ferritin using a glycine-serine (GS)-rich linker. The resulting Gn-ferritin (GnFt) nucleotide sequence was codon-optimized for expression in insect and mammalian cells, using GenSmart Codon Optimization (GenScript). For secretion into the culture medium, N-terminal signal peptides from Autographa californica nuclear polyhedrosis virus major envelope glycoprotein (GP67) and human interleukin-2 (IL2) were used for insect and mammalian cells, respectively. An alanine residue was added downstream the signal peptides, followed by a hexahistidine tag to facilitate purification, resulting in two constructs: GP67-A-GnFt and IL2-A-GnFt. These sequences were synthesized and cloned into the pUC-GW-Kan expression vector (Azenta).
The synthesized sequences were further cloned into two different ligation-independent cloning (LIC)-adapted expression vectors [29]. GP67-A-GnFt was inserted into pFastBac C-term [30] for expression in insect cells, resulting in pFastBac-GP67-A-GnFt. Similarly, IL2-A-GnFt was inserted into the pBacMam C-term [30, 31] for expression in mammalian cells, yielding pBacMam-IL2-A-GnFt. Site-directed mutagenesis was performed to generate constructs lacking the alanine residue, resulting in pFastBac-GP67-GnFt and pBacMam-IL2-GnFt. All plasmids were transformed into DH10B-T1 competent cells (Thermo Fisher Scientific) and purified using E.Z.N.A.® Plasmid DNA Mini Kit I (Omega Bio-tek).
Additionally, dsDNA coding for human cluster of differentiation 5 (hCD5) and bovine prolactin (bPRL) signal sequences were synthesized by IDT and used to replace the IL2-A sequence in pBacMam-IL2-A-GnFt using InFusion® cloning (Takara), generating the vectors pBacMam-hCD5-GnFt and pBacMam-bPRL-GnFt. Further modifications involved extracting the human phosphoglycerate kinase (PGK) promoter and the enhanced green fluorescent protein (EGFP) sequences from the pRRLSIN.cPPT.PGK-GFP.WPRE vector and inserting them downstream of the GnFt coding sequence in pBacMam-hCD5-GnFt using InFusion® cloning, resulting in pBacMam-hCD5-GnFt-PGK-EGFP. This modification was intended to assess mammalian transduction via fluorescence-based methods. Final vectors were transformed into Stellar™ competent cells (Takara) according to the manufacturer’s instructions and purified using the GeneJET Plasmid Miniprep Kit. Primer sequences used for LIC, mutagenesis, and InFusion® cloning are detailed in Table S1. Signal peptide and protein constructs, including their corresponding amino acid sequences, are provided in Table S2.
For bacmid generation by transposition [32], donor plasmids were transformed into DH10EmBacY (Geneva Biotech) or MAX Efficiency™ DH10Bac (Gibco) competent cells. Bacmids were extracted using reagents from the miniprep kits, followed by precipitation with ethanol/isopropanol, resuspension in water, and storage at -20 °C until further use.
DNA concentration was determined spectrophotometrically at 260 nm using a Nanodrop OneC (Thermo Fischer Scientific).
Cell lines and culture media
Insect cell lines Sf9 (Invitrogen), SuperSf9-2 (SuperSf9, OET), High Five™ (Hi5, Gibco), Hi5-derivatives Tnms42 [33, 34] and BTI-Tnao38 (Tnao38) [35, 36], and mammalian cell lines HEK293-6E (HEK293, NRC Canada) and FreeStyle™ CHO-S cells (CHO, Gibco) were passaged every 2–3 days in 125 mL–2 L shake flasks (Corning) with a working volume of 10–20%.
Insect cells were maintained at 27 °C in an Inova 44R shaking incubator (Eppendorf) set to 100 rpm using Sf-900™ II SFM (Sf900II) or Express Five™ SFM supplemented with 20 mM L-Glutamine (for Hi5 only). Mammalian cell lines were maintained at 37 °C in a humidified atmosphere with 5% CO2 in a Multitron shaking incubator (Infors HT) set to 100 rpm. HEK293 were grown in FreeStyle™ F17 Expression Medium supplemented with 4 mM GlutaMAX™, 0.1% Pluronic™ F-68 and 25 µg/ml Geneticin™, while CHO were cultivated in CD CHO Medium supplemented with 8 mM GlutaMAX™ (all from Gibco).
Viable cell density (VCD) and cell viability were assessed using the trypan-blue exclusion method with a Cedex HiRes cell analyser (Roche) following the manufacturer’s instructions.
Baculovirus generation, amplification and storage
Sf9 cells (2 mL) were seeded at 0.5 × 106 cells/mL in 6-well culture plates and allowed to adhere. For each transfection reaction, 8 µl of Cellfectin™ II (Gibco) and 1 µg of bacmid DNA were diluted in 200 µl of Sf9 medium and incubated at room temperature (RT) for 30 min. Medium in the culture plates was aspirated and replaced with 1.8 mL of fresh medium, along with the transfection mixture. After 96 h of incubation at 27 °C, the recombinant baculovirus (rBAC) P0 was harvested. For rBAC amplification, Sf9 cells were infected at 1 × 106 cell/mL using varying dilutions of virus. rBAC were harvested by centrifugation (200×g, 10 min + 2,000×g, 20 min, 4 °C) when viability reached 80–90%, typically after 3–4 days. rBAC stocks were amplified up to P2 and stored at 4 °C in the dark until further use.
For BacMam applications, certain rBAC stocks were concentrated using PEG precipitation method. Briefly, rBAC stocks were mixed with 20% (w/v) PEG 8000 solution to a final concentration of 2% (v/v). The mixture was incubated overnight on a roller mixer at 4 °C and centrifuged (3,200×g, 30 min, 4 °C). The resulting pellets were resuspended in PBS supplemented with 0.5 M sucrose using 1/10 to 1/100 of the starting volume.
Baculovirus infectious titers were determined by the MTT assay [37, 38].
Small-scale expression assay
Expression assays were conducted in triplicate using 125-mL shake flasks with a working volume of 10–20%. Tested conditions included different cell lines, signal sequences, cell concentration at transduction/infection, multiplicities of infection (MOIs, defined as the number of plaque forming units (pfu) of virus per number of cells), media exchange, sodium butyrate (NaBut) concentration and timing of addition, and temperature shift. Samples were collected every 24 h to monitor VCD, cell viability, and GnFt production via western blot.
Comparison of GnFt expression in mammalian cells using established conditions of BacMam expression
Mammalian cell cultures (HEK293 and CHO cells) were grown in triplicate in 50-mL (5 mL working volume) Corning® mini bioreactors and transduced at 1 × 106 cells/mL using rBAC containing the construct hCD5-GnFt-PGK-EGFP at three different MOIs: 2, 20, and 100 pfu/cell. Samples were collected every 24 h to monitor VCD, cell viability, and GnFt expression via western blot.
Purification using His•Bind resin
Sf9, Hi5, and HEK293 cultures were grown in 2 L (250 mL working volume) shake flasks using established optimal conditions. Cell cultures were harvested by centrifugation (2,000×g, 20 min) and filtered (0.22 μm). Clarified supernatants were concentrated and dialysed 5× using 20 mM Tris, 500 mM NaCl, pH 7.5 (base buffer) and a 1000 kDa Pellicon® 2 Cassette with a Ultracel® membrane (Millipore) operated in tangential flow filtration (TFF) mode. The resulting retentates were filtered (0.22 μm), supplemented with 10 mM imidazole (final concentration), and incubated with 1-mL His•Bind resin® (Millipore) for 1 h at 4 °C in a roller mixer. Protein was purified using Econo-Pac® gravity chromatography columns (Bio-Rad). For Sf9 and Hi5 cells, wash and elution were performed with base buffer supplemented with 70 and 250 mM imidazole, respectively. For HEK293, a step gradient ranging 25–250 mM was used.
Purification using ProteIndex™ Ni-Penta™ prepacked cartridges
Sf9 cells were grown in 2 L (200 mL working volume) shake flasks using established optimal conditions. Clarified supernatants were filtered (0.22 μm) and either directly loaded (DL) into 1-mL ProteIndex™ Ni-Penta™ agarose 6FF prepacked cartridges (Marvelgent Biosciences) or concentrated/dialysed by TFF as previously described. The resulting retentate was filtered (0.22 μm) to obtain the treated clarified (TC). Protein suspensions (DL and TC) were loaded at 1 mL/min onto a 1-mL ProteIndex™ cartridge, pre-equilibrated with base buffer, connected to an Äkta Explorer system (Cytiva). The column was washed with 20 mM imidazole, and elution performed using a 20-min linear gradient up to 250 mM imidazole.
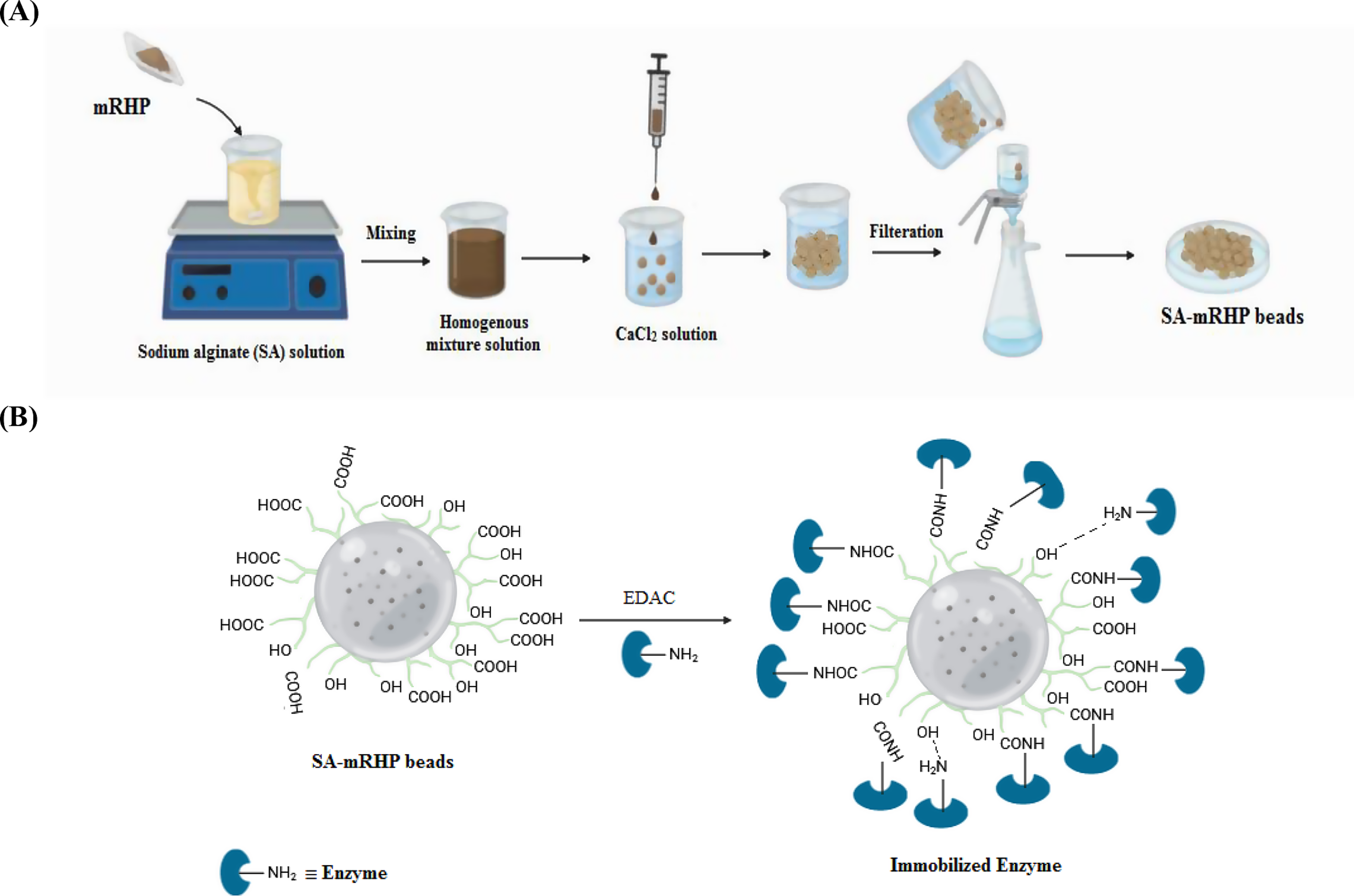
Production of ferritin nanoparticles devoid of antigen
The bullfrog-H. pylori hybrid ferritin (Ft) devoid of the Gn antigen contained a hexahistidine tag at the N-terminal and was synthesized and cloned into pOET1.1 (Oxford Expression Technologies) by GenScript. rBAC containing the Ft sequence was generated by homologous recombination using flashBAC ULTRA (Oxford Expression Technologies) as per manufacturer’s instructions, and amplified as described above. Sf9 cultures were grown in 2 L (200 mL working volume) shake flasks and infected at 2 × 106 cell/mL and MOI of 1 pfu/cell. After 72 h, cell pellets were harvested by centrifugation and resuspended in base buffer supplemented with 0.2% Deviron® C16 (Merck), 50 U/mL Benzonase® (Millipore), 2 mM MgCl2, and cOmplete ™ protease inhibitor cocktail (Roche) using 1/10 of the starting volume. The suspension was incubated on a roller mixer for 30 min at 4 °C, followed by centrifugation (16,000×g, 1 h, 4 °C). The lysate was filtered (0.45 μm) and purified using a 1-mL ProteIndex™ cartridge as described above.
Protein storage and quantification
After chromatography, GnFt- and Ft-containing fractions were pooled, concentrated, dialyzed against formulation buffer (20 mM Tris, 150 mM NaCl, pH 7.5) using 100-kDa cutoff Amicon® centrifugal units (Millipore), filtered (0.22 μm), and stored at -80 °C. Protein concentration was estimated using Pierce™ BCA protein assay kit (Thermo Scientific) or absorbance at 280 nm using the extinction coefficient 4.74 × 104 M− 1cm− 1 (GnFt, 1-mer) and 2.14 × 104 M− 1cm− 1 (Ft, 1-mer), as estimated by the ProtParam tool (Expasy [39]), in a Nanodrop OneC.
SDS-PAGE and western blot analysis
Samples were mixed with LDS sample buffer and sample reducing agent, heated (3 min, 99 °C), loaded onto 4–12% bis-tris gels, and electrophoresis performed (40 min, 200 V, 400 mA) using MES SDS running buffer and SeeBlue™ Plus2 pre-stained protein standard. Proteins were transferred onto nitrocellulose membranes using iBlot™ 2 transfer stacks and gel transfer device (10 min, 20 V) (all from Invitrogen).
After transfer, membranes were blocked for 1 h at RT in tris-buffered saline (Sigma-Aldrich) with 0.1% Tween-20 (Millipore) (TBST) containing 5% (w/v) skim milk (Millipore). Membranes were incubated overnight at RT with primary antibodies, followed by a 1 h incubation at RT with secondary antibodies; membranes were washed three times with TBST for 5 min between each incubation step. Bands were revealed using NBT/BCIP 1-Step (Thermo Fisher Scientific) and documented with an iBright FL1500 (Invitrogen).
Antibodies were prepared in blocking solution and included mouse monoclonal anti-RVFV Gn glycoprotein (NR-43185 [40], BEI Resources) and mouse 6x-HisTag monoclonal antibody (MA1-21315, Invitrogen) as primary antibodies (1:1,000–2,000) and goat anti-mouse IgG conjugated with alkaline phosphatase (A3438-25ML, Sigma-Aldrich) (1:5000) as secondary antibody.
Densitometry
Densitometry analysis of protein bands on the western blot membranes was conducted using ImageJ software [41]. Relative volumetric titer for each sample was calculated by dividing the band intensity area of that sample by the intensity area of a given condition. Relative cell-specific productivity was determined similarly, with the intensity area further normalized by dividing it by the area under the VCD curve.
High performance liquid chromatography (HPLC)
Samples were analysed on a HPLC system with a Vanquish Diode Array Detector (Thermo Scientific) using a Bio SEC-5 column (500 Å, 5 μm, 4.6 × 300 mm, Agilent) at a flow rate of 0.3 mL/min. Formulation buffer was used as mobile phase and BEH450 SEC Protein Standard Mix (Waters) as molecular weight standard.
Dynamic light scattering (DLS)
The size distribution of particles in protein samples was estimated by DLS using a Zetasizer Ultra (Malvern Panalytical). Protein samples were diluted in formulation buffer to a final volume of 800 µL and placed in disposable cuvettes. Each sample was measured three times, and data analysed using ZS Xplorer software (Malvern Panalytical).
Mass photometry
The molecular mass of proteins was determined using the mass photometer SamuxMP (Refyne). Formulation buffer (18 µL) was added to the slide and autofocus adjusted. Subsequently, 2 µL of sample was resuspended in the buffer droplet. Data was acquired and analyzed using AcquireMP and Discover MP software (Refyne), respectively.
Negative staining transmission electron microscopy (nsTEM) and single-particle analysis
Sample preparation and imaging of nanoparticles by nsTEM were performed at the Electron Microscopy Facility at CIC bioGUNE. Purified proteins were prepared at 0.1–0.3 mg/mL in formulation buffer. Freshly glow-discharged CF400-Cu grids (EM Sciences) were incubated with an 8-µL droplet of each sample for 60 s and then transferred to an 8-µL MilliQ water droplet for 30 s. Grids were subsequently placed on a uranyl acetate droplet for two consecutive 30 s incubations, and left to dry prior to visualization; excess liquid was removed using Whatman paper after each incubation.
For imaging, grids were transferred into an EM-11,170 pentaholder (Gatan) and visualized on a JEM-1230 transmission electron microscope (JEOL) equipped with a LaB6 emission gun (FEG), operated at 100 kV, and spherical aberration of 2.0 mm. Digital images were recorded on a 4 K×4 K (15 μm pixels) Ultrascan4000™ charge-coupled device camera (Gatan) using a nominal magnification of 40,000 × (2.9 Å/pixel) with a total dose on the order of 40–60 electrons/Ų per exposure, at defocus values ranging from 3.0 to 5.0 μm.
Single-particle analysis was performed using CryoSPARC v4.3 [42]. From 35 micrographs, 131,071 and 47,020 particles were extracted for Ft and GnFt, respectively, using a template picker. After four rounds of 2D classification, the final 2D classes contained 10,820 Ft and 2,888 GnFt nanoparticles.
Biolayer interferometry (BLI) of antibodies binding to nanoparticles
Nanoparticle binding to Gn-specific monoclonal antibodies was measured with an Octet RED96 (FortéBio). NTA biosensors (Sartorius) were equilibrated in kinetic buffer (Sartorius) for 180 s, loaded with GnFt or Ft nanoparticles (10 µg/mL) for 300 s, and subjected to three-fold serial antibody dilutions (starting at 90 nM) for 600 s association and 600 s dissociation in kinetic buffer. Regeneration between experiments was achieved with glycine (100 mM, pH 2.0) followed by nickel recharging with NiSO4 (100 mM). Kinetic constants were determined by fitting data to a non-linear regression model in GraphPad Prism v10.4.2. Antibodies included monoclonal anti-RVFV Gn NR-43185, NR-43189, NR-43190, and NR-43195 (BEI Resources).
Statistical analysis
Statistical analysis and graphical representation were performed using GraphPad Prism v10.4.1. To determine statistical significance, unpaired t-test was used for comparisons between two groups. For multiple groups, one- or two-way analysis of variance (ANOVA) followed by Tukey’s or Šídák’s multiple comparisons test was employed. A p-value greater than 0.05 was considered non-significant (ns). Significance levels are indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Results are displayed as mean ± standard deviation (SD).





Comments (0)