
Remember me
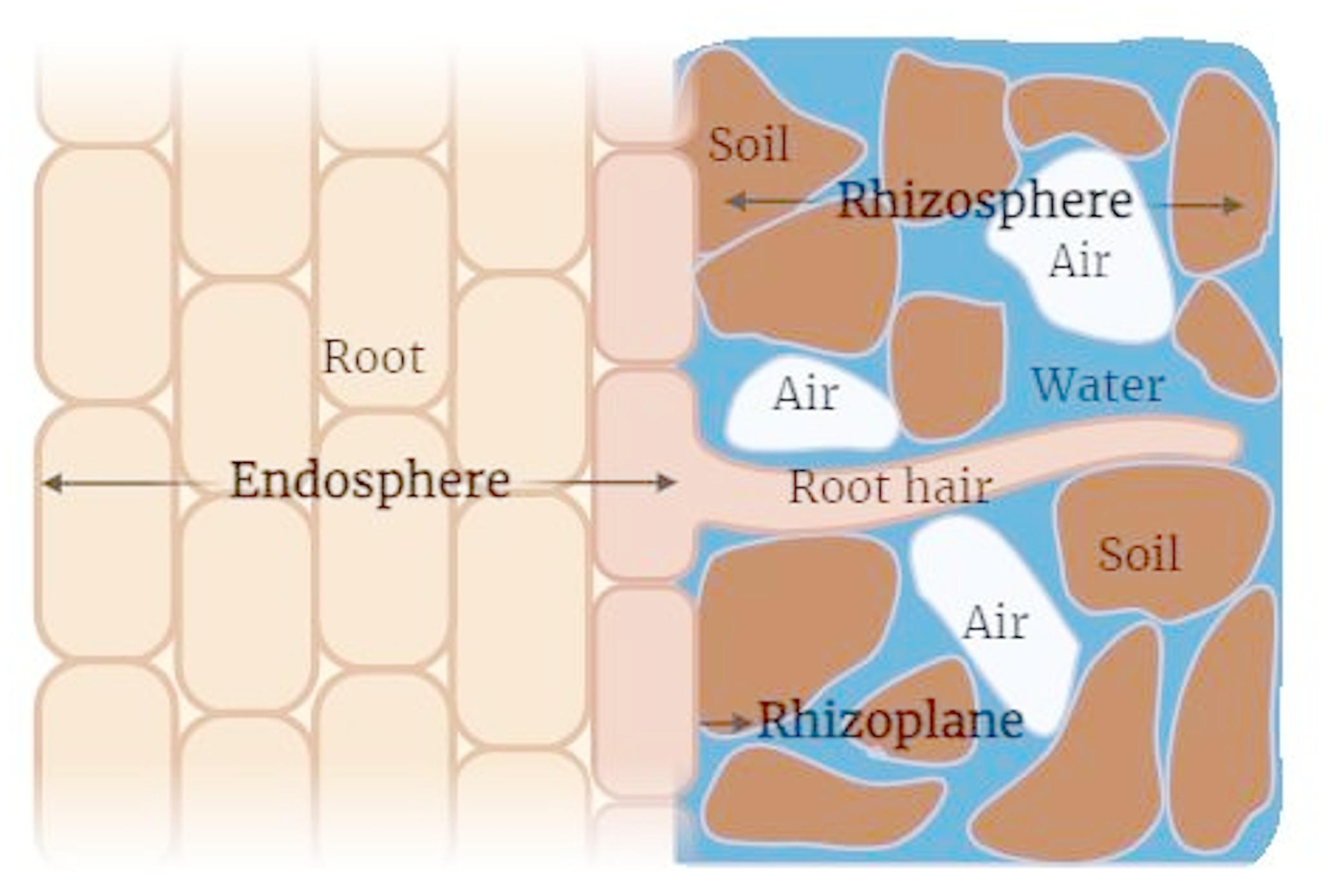

Rhizospheric soil samples (Table 1) were collected in January 2022 from eight different Egyptian wheat genotypes; representing both wild and cultivated genotypes. The wild genotypes are represented by Triticum dicoccum and Triticum monococcum, while the cultivated genotypes were Giza 168, Gemiza 9, Gemiza 11, Masr 1, Sids 13, and Shandweel 1. These cultivated genotypes represent the most promising wheat genotypes developed by the Agricultural Research Center (ARC), Giza, Egypt. The trial was conducted at the experimental field station of the Agricultural Genetic Engineering Research Institute (AGERI), ARC, in Giza, Egypt, with coordinates 30°01’25” N, 31°12’11” E.
Five specimens of each wheat variety were collected aseptically and brought to the laboratory in sterile bags. The root samples were taken from a depth of 30 cm, and the adhering soil was manually shaken or loosened from the roots. The roots were cut with sterilized scissors and placed in tubes containing 10 mL of sterile 1% NaCl solution to dissolve the microbes associated with the rhizosphere by shaking. The resulting suspension was vortexed to further release rhizosphere bacteria. Plant debris and roots were then removed, and the solution was filtered through a sterile 100 μm cell strainer.
The filtrate was centrifuged at 3000×g for five minutes, the supernatant was discarded, and the bacterial pellet was resuspended in 1% NaCl solution (McPherson et al. 2018). The samples were then stored at − 20 °C prior to DNA extraction. Genomic DNA was extracted using the PowerSoil DNA Isolation Kit (Qiagen, Cat. No. 47014) according to the manufacturer’s recommended protocol (Fouad et al. 2025).
Table 1 Library names, corresponding Egyptian wheat genotypes, treatment conditions (control or drought), and their respective accession numbersMethodsMetagenomic amplicon sequencingTargeted amplicon sequencing of the 16 S rRNA gene was performed to characterize the bacterial communities in the rhizosphere of wheat. The hypervariable V3–V4 regions of the 16 S rRNA gene were amplified with region-specific primers: 341 F (5’-CTACGGGGNGGGCWGCAG-3’) and 806-R (5’-GGACTACNNGGGTATCTAAT-3’). Sequencing was performed on Illumina’s NovaSeq platform (Illumina, San Diego, CA, USA) according to the manufacturer’s protocols, which enables deep sequencing of amplified fragments at high throughput.
Sequence analysisData analyses were performed using the bioinformatics software package QIIME2 (www.qiime2.org) via the q2cli interface. Forward and reverse sequences were merged, denoised, dereplicated and filtered for chimaeras using the DADA2 plugin (q2-dada2). Amplicon sequence variants (ASVs) were aligned using MAFFT (via q2-alignment) and a phylogeny was generated using FastTree (via q2-phylogeny). Based on a preliminary analysis, a subsampling depth of 2000 was chosen.
Taxonomic classification of ASVs was performed using the 99% Greengenes 13.8 reference database (q2-feature-classifier), removing unwanted taxa such as those containing mitochondria, archaea, eukaryotes or chloroplasts in their taxonomic annotation. For visualization, taxa bar plots and collapsed tables were created and the data for each taxa level was summarized in a table. For additional analysis, R programming was used with the output files generated by QIIME2.
The package QIIME 2R enabled the import of QIIME artifacts into an R session. The package phyloseq was used to import, store, analyze, and visually display complex phylogenetic sequencing data organized into Operational Taxonomic Units (OTUs), especially when associated specimen data, a phylogenetic tree, and taxonomic assignments of the OTUs were available. The package vegan was used for diversity analysis, while the packages ggplot2, pheatmap and RColorBrewer were used to create bar charts and heatmaps for visualization.
Visual exploration of the taxonomic composition of the studied rhizosphere community was performed by comparing abundances. The taxonomic composition was considered at phylum, class and family levels, categorized according to an experimental factor that grouped the experiment into four categorical aspects: rhizosphere soil samples of the cultivated genotypes under controlled conditions, rhizosphere soil samples of the drought- treated cultivated genotypes, rhizosphere soil samples of the wild genotypes under controlled conditions and rhizosphere soil samples of the drought- treated wild genotypes.
Taxa were categorized into the categorical group based on the total sum of their counts, with the top taxa (n = 12) displayed to show the percentage abundance. Alpha diversity analysis was performed using the Microbiome Analyst tool (available at: http://www.microbiomeanalyst.ca) (Dhariwal et al. 2017). Observed alpha diversity was used to quantify species richness; the richness (actual number) of unique taxa observed was calculated by the index Observed. In addition, evenness (number of abundances) was calculated using the Shannon, Simpson and Fisher’s indices to represent the true diversity of the community (Iannucci et al. 2021).
Beta diversity analysis was performed using the phyloseq package in R to compare the composition of the microbial community in the different samples. Bray-Curtis distance was used to calculate dissimilarities based on the presence, absence, and abundance of taxa. Principal coordinate analysis (PCoA) served as the ordination method, using the Bray-Curtis index as the distance metric. Statistical differences between groups were assessed using PERMANOVA to detect significant variations in microbial composition.
Differential abundance analysisThe taxonomic counting matrix at the phylum and class level, together with the corresponding metadata file, was imported into the iDEP (Integrated Differential Expression and Pathway Analysis) software to perform differential frequency analysis using limma-voom (Ge et al. 2018). This analysis aims to identify highly significant taxa that showed a distinct presence and contributed significantly to the observed differences between the control and drought groups.
These heat maps provided an intuitive and informative representation of the taxonomic composition of the samples and enabled the identification of patterns and relationships between taxa. The resulting heatmap, created using ClustVis software (Metsalu and Vilo 2015), was subjected to further analysis and interpretation to identify meaningful relationships between the samples and patterns of taxonomic composition. This comprehensive analysis enabled a deeper understanding of taxonomic dynamics and their relationships to control and drought conditions.
Core MicrobiomeIn our study, we performed a core analysis of the microbiome to identify taxa or traits that are stable in the composition of the microbial community. This approach helps to understand the basic components that are stable within the microbiota. We determined two central parameters with the core microbiome analysis.
The first parameter, called sample prevalence, defines the minimum proportion or percentage of samples in which a taxa or trait must be present to be considered part of the core microbiome.
A value of 20% was set as the threshold for sample prevalence. The second parameter, relative abundance, defines the proportion of the occurrence of a taxa or trait in the samples that qualifies it as a core member. In our analysis, a relative abundance threshold of 0.01% was used. By using web-based Microbiome Analyst tool (available at: http://www.microbiomeanalyst.ca).
The analysis of the core microbiome was performed using the core function from the R package ‘microbiome’. This function enables efficient determination and visualization of core taxa or features.
Correlation analysisA correlation analysis was performed to determine whether certain traits showed different patterns in the cultivated and wild genotypes under different conditions, i.e. drought and normal irrigation. Correlation was performed at the level of strain, class and family taxonomy. The Pearson r correlation method was used as a measurement distance to determine whether a linear relationship exists between two taxa. The MicrobiomeAnalyst tool was used for this purpose (available at: http://www.microbiomeanalyst.ca) (Dhariwal et al. 2017).
Machine learning-based functional profilingThe 16 S rRNA sequencing data were used to estimate the functional profiles of the microorganisms in the wheat rhizosphere by inferring their traits based on their genetic relatedness to fully sequenced and annotated organisms. This process was carried out using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt13). Gene family frequencies were generated for each sample using the KEGG database (KO).
We then used the Shotgun Data Profiling (SDP) module in MicrobiomeAnalyst 2.0 to categorize the predicted metagenomes into broader functional categories and link them to metabolic pathways. We then used supervised Random Forests (RF), a classification algorithm consisting of unpruned decision trees created from training data. These trees were created using a subset of OTUs randomly selected from a bootstrap sample. The RF classifier was built by creating 5,000 classification trees and the prediction performance was evaluated using out-of-bag cross-validation and confusion matrices.
To identify the most predictive taxa for each microbiome assemblage, we determined the mean decrease in accuracy using the importance matrix. The Random Forest algorithm in MicrobiomeAnalyst 2.0 was used. This approach identified the OTUs that differed most from the different soils based on the decrease in model accuracy by leave-one-out cross-validation.
Results Microbiome profilingTo assess the diversity of the wheat rhizosphere microbiome, we performed alpha, and beta diversity analyzes. Through taxonomic classification, a total of 10,858 Operational Taxonomic Units (OTUs) were identified in the selected Egyptian wheat genotypes. After eliminating unwanted taxa, as previously mentioned, we classified and categorized the remaining 9,410 OTUs into seven taxonomic ranks, resulting in 356 OTUs [S1].
To determine the relative abundance of the different microbial groups associated with wheat plants, we performed a taxonomic analysis at the level of strains, classes and families. At the strain level, we observed four dominant and abundant phyla among the identified OTUs. The phyla Proteobacteria showed a prevalence of 48% under drought conditions for both wild and cultivated genotypes, with a slightly higher presence (58% and 53%) under control conditions for wild and cultivated genotypes, respectively (Fig. 1).
Fig. 1
Drought- and control-induced taxonomic composition of the wheat rhizosphere microbiome. Relative abundance at the (A) phylum, (B) class and (C) family levels. Stacked bar graphs showing the taxonomic composition of the wheat rhizosphere microbiome under drought conditions (blue) and under control conditions (red). Only the 12 most abundant taxa are shown, with relative percentages. Common taxa accumulate in response to drought, including Actinobacteria and Verrucomicrobia, and the changes in microbial communities in response to drought are determined by changes in the abundance of these taxa
Bacteroidetes were represented at 19% and 20% in the wild and cultivated genotypes under drought conditions, respectively, and maintained similar proportions in both genotypes under control conditions (16 and 21, respectively). Actinobacteria were detected at 14% and 13% in the wild and cultivated genotypes under drought conditions and at 7% and 6% under control conditions, respectively. Finally, Verrucomicrobia was present at 4% and 5% in the wild and cultivated genotypes under drought conditions, respectively, and showed a higher percentage of 6% in both genotypes under control conditions.
The relative abundance of these phyla provided information on the composition of the microbial community associated with the wheat plants (Fig. 1A). A detailed class-level analysis assigned a substantial proportion of the Operational Taxonomic Units (OTUs) to specific classes. Gammaproteobacteria accounted for 27% of the taxa present under drought conditions for wild and cultivated genotypes, and 36% and 31% under control conditions, respectively.
Alphaproteobacteria were present in 12% and 15% of wild and cultivated wheat genotypes respectively, under drought conditions and 13% and 15% under control conditions. Cytophagia accounted for 11% and 12% under drought conditions and 8% and 10% under control conditions for the wild and cultivated genotypes, respectively. Bacilli were present at 4% and 3% under drought conditions and 3% and 2% under control conditions for the wild and cultivated genotypes, respectively. These results emphasize the prevalence of these specific classes under different conditions and shed light on their dynamics within the microbiota.
These classes accounted for a significant proportion of the taxonomic diversity within the wheat-associated microbial community (Fig. 1B). Microbial composition at the family level was analyzed under both control and drought conditions in the rhizosphere of wild and cultivated wheat genotypes. Pseudomonadaceae was the predominant family in these analyzes. Under controlled conditions, the rhizosphere of the wild wheat genotypes showed a higher prevalence of Pseudomonadaceae (26%) than that of the cultivated genotypes (20%). Under drought conditions, the prevalence was 15% for both genotypes (Fig. 1C).
The analysis of alpha diversity at the species level, using the Kruskal-Wallis non-parametric statistical method, provided valuable insights into the diversity of the microbial community. The observed alpha diversity, which represents the actual species richness (Fig. 2A) showed no significant difference between the compared groups (p-value = 0.57059), indicating random variation in the observed number of species. However, the Simpson diversity index and the Shannon diversity index showed notable differences.
Fig. 2
Alpha Diversity of the Microbiome in the Wheat Rhizosphere. Observed alpha diversity (species richness) using the Observed index (A), Shannon diversity index (B) and Simpson diversity index (C). Microbial diversity across experimental groups: control versus drought (n = 4) indicated by boxplots Shannon and Simpson indices of the respective microbial diversity show that microbial diversity was distinct between conditions (P < 0.005), suggesting shifts in community evenness and taxonomic richness. Typed statistical significances were determined using Kruskal-Wallis test
Fig. 3
Principal coordinate analysis (PCoA) based on Bray-Curtis distance to visualize the beta diversity of the rhizosphere microbiome of wild and cultivated wheat genotypes under control and drought conditions. Distinct cluster patterns indicate shifts in the microbial community in response to genotype and environmental stress. The PERMANOVA test showed significant differences (p = 0.001 to 0.0285) between groups, with the highest significance observed in the cultivated genotypes. See Table 2 for detailed statistics
The Simpson index (p-value = 0.027147) (Fig. 2C) showed significant differences indicating variations in species diversity and evenness within the microbial communities. The Shannon index (p-value = 0.039408) also emphasized the different distribution and abundance of species in the groups (Fig. 2B). These results emphasize the importance of considering both abundance and evenness when assessing microbial diversity, as shown by the significant results of the Simpson and Shannon diversity indices at the species level.
Using Bray-Curtis dissimilarity analysis and weighted UniFrac analysis, we aimed to assess beta diversity and investigate the variation in microbial community composition within these genotypes. Bray-Curtis dissimilarity analysis showed similarities in microbial community composition between the control and drought-treated wheat genotypes Fig. (3). Principal component analysis revealed that PC1 explained 42.8% of the variance and PC2 explained 30.5% of the variance. These differences between the control and drought groups were not statistically significant, indicating a comparable microbial community composition.
However, the weighted UniFrac analysis revealed significant dissimilarities indicating differences in the phylogenetic composition of the microbial communities. These results provide valuable insights into the effects of drought on wheat-associated microbial communities and emphasize the role of phylogenetic relationships in shaping the observed dissimilarities (Fig. 3). The pairwise PERMANOVA test showed higher significance when comparing control and drought conditions among the cultivated genotypes, with a p-value of 0.001 (FDR-adjusted p = 0.006). This was followed by significant differences between other pairwise comparisons between wild and cultivated genotypes under drought (Table 2).”
Table 2 Statistical comparison of microbial community structures between drought and control genotypes for beta diversity analysisComparative assessment of microbial abundance and correlation analysisThe differential abundance analysis revealed different taxa whose abundance varied considerably between the two groups. On the other hand, correlation analysis investigated relationships and associations between taxa and different variables such as drought conditions and controlled environments.
Differential abundance analysisDifferential abundance analysis was performed to identify microbial taxa that exhibited significant differences in abundance between different conditions or groups within the microbial community. For this analysis, the limma voom test implemented in the iDEP software was used. Heatmap clustering was then performed using ClustVis. At the phylum level, the analysis revealed several highly significant and abundant phyla that showed notable differences between the control and drought-treated groups (Fig. 4). The false discovery rate (FDR) threshold was set at less than 0.05%.
Fig. 4
Hierarchical clustering heatmap of the differently abundant microbial taxa at phylum and class level. The color scale is based on relative abundance (from red for high abundance to blue for low abundance). Valuable taxa such as OD1, WS2, Chlorobi, ABY1 and SHA-109 show a clear difference between drought and control conditions
The identified phyla were OD1 (FDR = 3.41 × 10− 6), WS2 (FDR = 1.45 × 10− 5) and Chlorobi (FDR = 0.00549). In addition, the class-level analysis revealed highly significant and frequent classes that showed a significant difference between the control and drought-treated groups, also at an FDR threshold of less than 0.05%. These classes were ABY1 (FDR = 6.72 × 10− 8), Actinobacteria (FDR = 1.45 × 10− 5), SHA-109 (FDR = 1.45 × 10− 5), ZB2 (FDR = 9.05 × 10− 5), Rhodothermi (FDR = 0.00419), Verrucomicrobiae (FDR = 0.00419) and Pedosphaerae (FDR = 0.02398) (Fig. 4).
Correlation analysisIn our correlation analysis, we investigated the relationship between the abundance of phyla and the different conditions in the rhizosphere of wild and cultivated wheat genotypes. Three significant phyla, Actinobacteria, OD1 and Chlorobi, were identified based on their respective false discovery rate (FDR) values. Actinobacteria (FDR = 0.00028302) showed a positive correlation with the drought-treated wild genotypes, indicating higher abundance.
Interestingly, OD1 (FDR = 0.0012082) showed a negative correlation with both the wild and cultivated wheat genotypes under drought conditions, but showed a contrasting positive correlation under control conditions, indicating higher abundance. Similarly, Chlorobi (FDR = 0.024999) showed a negative correlation with the drought conditions but exhibited a positive correlation, indicating higher abundance under the control conditions for both the wild and cultivated genotypes (Fig. 5A, B, C) (Table 2). At the taxonomic class level, we identified five significant taxa: Actinobacteria, Verrucomicrobiae, ABY1, Bacteroidia and TA18, based on their respective FDR values (0.00052714, 0.00052714, 0.00065193, 0.043198, 0.043198).
Fig. 5
The significant microbial taxa (phylum, class and family level) with correlation patterns between drought and control are shown in boxplots. (A-C) significant phylum-level correlations to each other (Actinobacteria, OD1, Chlorobi). (D-H) significant class-level correlations to each other (Verrucomicrobiae, ABY1, Actinobacteria, Bacteroidia, TA18). (I-T) significant family-level correlations (eg. Streptomycetaceae, Verrucomicrobiaceae, Nocardioidaceae, Methylobacteriaceae). FDR-adjusted Pearson correlation values were calculated and applied
Figure 5A, Actinobacteria showed a negative correlation with the cultivated and wild genotypes under control conditions, whereas under drought conditions they showed a contrasting positive correlation in both sample types, indicating higher abundance. Conversely, Verrucomicrobiae, ABY1, Bacteroidia and TA18 showed a positive correlation with the control genotypes, indicating higher abundance, and switched to a negative correlation under drought conditions (Fig. 5D, E, G, H) (Table 2).
We identified 12 significant taxa at the family level. The FDR was used to test the reliability of these correlations. Interestingly, the Streptomycetaceae showed a significantly low false discovery rate (FDR) of 1.49E-05. Moreover, under control conditions, it showed a consistent negative correlation with both cultivated and wild genotypes. However, under drought conditions, it showed a strong positive correlation and reached its highest abundance.
The Verrucomicrobiaceae, on the other hand, showed an FDR of 0.00087393, indicating significant correlations. This family showed a positive correlation with the cultivated genotypes under control conditions and the drought- treated wild genotypes. Conversely, it showed a negative correlation under the opposite conditions. The Nocardioidaceae showed an FDR of 0.0023626, also indicating a significant correlation. This family showed a positive correlation and reached its highest abundance under drought conditions in both cultivated and wild genotypes.
Promicromonosporaceae and Nocardiaceae, both with an FDR of 0.0054741, showed consistent negative correlations under control conditions and positive correlations under drought conditions, with the latter showing the highest abundance. Brucellaceae, despite their very low abundance, showed an FDR of 0.0079671, indicating significant correlations, especially a positive correlation under drought conditions, which stands out in terms of abundance.
Sphingomonadaceae, with an FDR of 0.023311, showed a positive correlation, and reached their highest abundance in the control cultivated genotypes, but showed a negative correlation under the drought-treated conditions. Cystobacteraceae and Chromatiaceae both showed an FDR of 0.028592, indicating significant correlations. Cystobacteraceae showed a positive correlation under drought conditions and reached its highest abundance, while Chromatiaceae showed a positive correlation under control conditions and also reached its highest abundance.
Finally, Glycomycetaceae, Methylobacteriaceae and Xanthomonadaceae, all with an FDR of 0.030596, showed significant correlations. Glycomycetaceae showed a positive correlation under drought conditions and reached its highest abundance, while Methylobacteriaceae and Xanthomonadaceae showed a positive correlation with the highest abundance under control conditions (Fig. 5I, J, K, L, M, N, O, P, Q, R, S, T) (Table 3).
Table 3 Correlations at taxa level with environmental conditions (control vs. drought)Core MicrobiomeWe defined the core microbes based on their prevalence in all analyzed rhizosphere genotypes (prevalence = 1) and their relative abundance of more than 0.01%. The result of this analysis was presented in the form of a heat map where the Y-axis shows the prevalence of core features in the detection limit (relative abundance), which is plotted on the X-axis.
In Figure (6), we present the results of the analysis of the core microbiome using a heat map that provides a clear visualization of the prevalence of core features across the spectrum of relative abundance. This visualization improved our understanding of the stable components within the microbial community and their importance in shaping the overall ecosystem. Four major phyla consistently met these criteria and together accounted for 23.5% of the observed phyla in the wheat rhizosphere. Among these major phyla, Verrucomicrobia, Proteobacteria, Bacteroidetes and Actinobacteria stood out as dominant taxa, emphasizing their significant presence and potential role in the rhizosphere ecosystem (Fig. 6A).
Fig. 6
Heatmap of the composition of the core microbiome under drought and control conditions. (A) Core microbiome at phylum level, (B) core microbiome at class level, (C) core microbiome at family level. The persistently high prevalence of core taxa, especially Proteobacteria, Actinobacteria, and Verrucomicrobia, also highlights the fundamental importance of these taxonomic groups for the wheat rhizosphere ecosystem. Detection thresholds: 20% sample prevalence, relative abundance of 0.01%
At the class level, however, 13.5% of the total taxa observed were assigned to five key classes: Gammaproteobacteria, Cytophagia, Betaproteobacteria, Alphaproteobacteria and Actinobacteria. These classes were identified as key components due to their uniform prevalence in all rhizosphere genotypes (Fig. 6B). In addition, Pseudomonadaceae and Cytophagaceae were consistent at the family level (Fig. 6C).
Functional prediction analysisPhylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to estimate the functional potential resulting from the information on the 16 S rRNA gene (Fig. 7). This approach allowed the inference of microbial functional profiles based on their genetic proximity to fully sequenced and annotated organisms. The results provided a table of gene family frequencies for each sample annotated via the KEGG database (KO) [S2].
Fig. 7
Stacked area chart showing the relative abundance of predicted metagenomic functions (KEGG pathways) in the wheat rhizosphere microbiomes under drought and control conditions using PICRUSt analysis. The Y-axis represents the relative abundance of the functional categories, indicating shifts in microbial metabolic potential
Fig. 8
Predictive functional genes of drought-treated and control genotypes, analyzed with Random Forest (RF). (A) Mean Decrease Accuracy (MDA) ranking of the 15 most predictive KEGG orthologs (KOs). (B) Classification importance (scaled) of various key microbial functional genes. RF identified K07133 (ATP-binding protein of the AAA + superfamily) as the most predictive feature that may be involved in microbial stress adaptation under drought conditions. The out-of-bag (OOB) error rate (14.2%) and the area under the curve (AUC) score (0.91) indicate a high classification accuracy
A graph was created showing the frequency of gene families for each sample, with gene families annotated using the KEGG database (KO) (Fig. 8). In addition, the predicted metagenomes were categorized into broader functional groups using RF analysis and linked to metabolic pathways to improve the understanding of functional potential within the microbial community.
The main finding of this analysis is that the predicted metagenomic functions in the wheat rhizosphere microbiome show significant shifts under drought conditions. The analysis revealed a set of fifteen KOs with high predictive significance, including K07133, K00525, K01507, K03413, K13525, K00600, K00772, K09013, K03636, K07151, K01704, K00762, K01840, K03709 and K01703. The identification of K07133 (uncharacterized protein) as the only significantly associated KO with drought-treated genotypes suggests a unique microbial functional adaptation or stress response that does not occur under normal conditions.
This KO, recognized as an uncharacterized protein comprising an ATP-binding protein, ATPase, AAA + superfamily, showed the highest decrease in mean value, indicating its significant predictive influence in the studied microbiome (Fig. 8).
In contrast, the other 14 KOs were predominantly associated with control genotypes, suggesting that metabolic pathways related to nucleotide metabolism (ribonucleoside diphosphate reductase, pyrE), energy metabolism (inorganic pyrophosphatase, Glycine hydroxymethyltransferase), stress response (Fe-S cluster assembly ATP-binding protein) and protein processing (transitional endoplasmic reticulum ATPase, glycosyltransferase) may be more active or stable under non-stressed conditions. This suggests that drought stress selectively alters microbial functional potential, possibly leading to the suppression of certain metabolic activities while promoting specific adaptations.
Comments (0)