Cell lines
The CT26, B16-F10 and ID8 cell lines were acquired from the American Type Culture Collection (ATCC). They were maintained in RPMI 1640/DMEM (HyClone) media supplemented with 10% fetal bovine serum (FBS) (HyClone) at 37 °C. Subsequently, ID8-luc cell lines were generated using viral transduction followed by selection with puromycin (1 µg/mL). Similarly, ID8 cells expressing green fluorescent protein and ovalbumin gene (ID8-EGFP-OVA) were generated using the same method. All cell lines were confirmed to be mycoplasma-free.
Preparation of bone marrow-derived macrophages (BMDMs)
Flushing of femurs and tibias of mice using DMEM was conducted to extract bone marrow cells. The resulting cell suspension was filtered using a 70 μm filter, then washed twice with PBS. The obtained cells were then cultured in complete medium containing 30% L929 cell supernatant. BMDMs were collected for experimentation on day 5.
Plasmid, virus and CAR-Ms
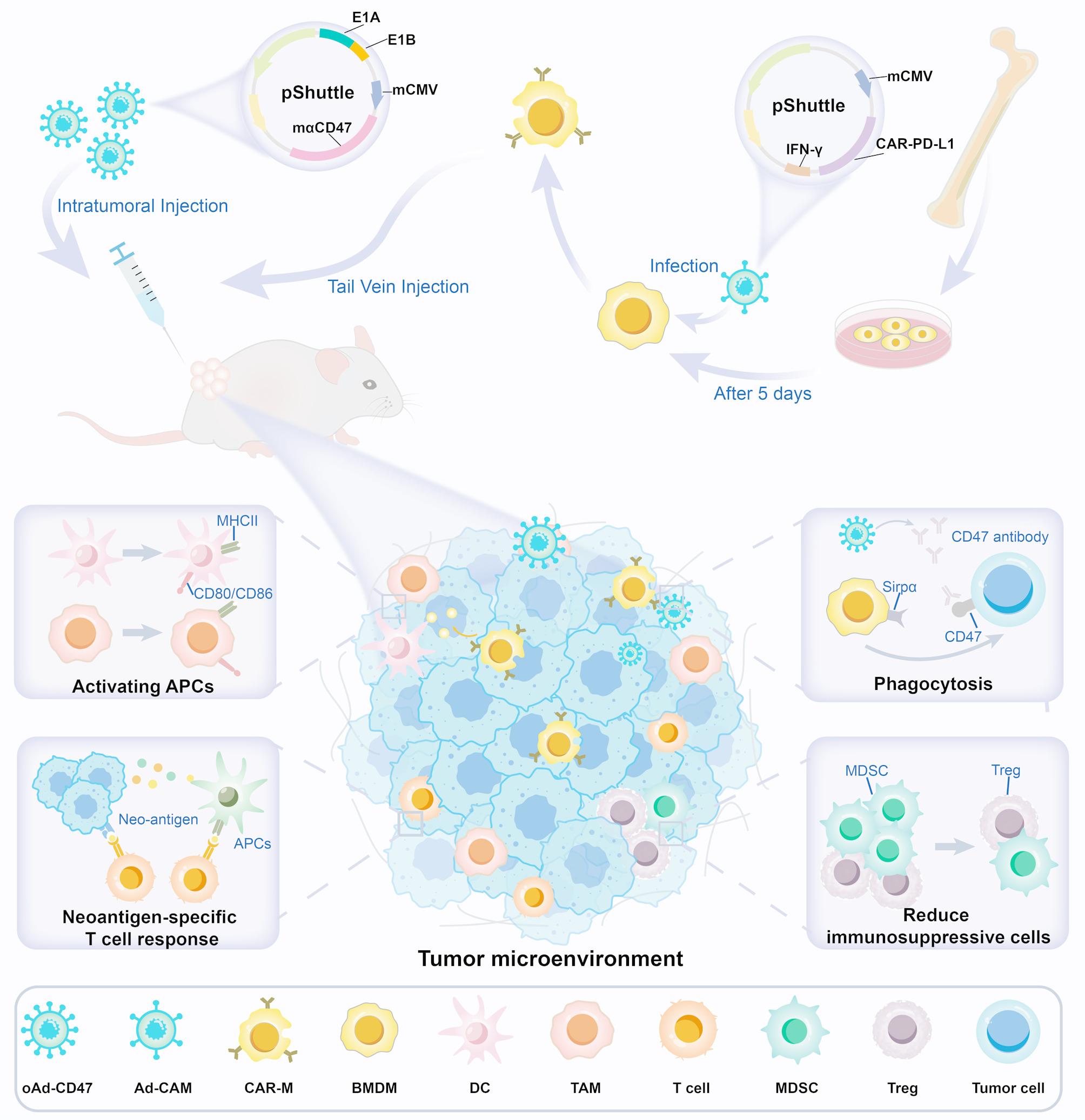
The replication-deficient adenoviral shuttle vector (pDC316-mPD-L1nb-CAR) was synthesized by Genewiz. HEK293A cells were co-transfected with shuttle vector containing CAR-mPD-L1-IFN-γ structure and a backbone plasmid for packaging the non-replicative adenovirus (Ad-CAM). The empty control vector (Ad-ON) retained the same backbone but lacked the CAR-mPD-L1-IFN-γ insert. Viral particles were amplified in HEK293 cells and purified by CsCl gradient centrifugation followed by dialysis using dialysis buffer. The adenoviral titer was determined using 50% tissue culture infection volume (TCID50) method.
The oncolytic adenovirus (oAd-CD47) was constructed as previously described [24]. The E1A-IRES-E1B gene was inserted into pDC316 under the control of the human telomerase reverse transcriptase promoter to construct an oncolytic adenovirus shuttle vector (pDC316-hTERT-E1A-IRES-E1B). The transgene mCD47nb-Fc was cloned into the aforementioned oncolytic adenovirus shuttle vector under the control of the mCMV promoter (pDC316-hTERT-E1A-IRES-E1B-mCD47nb-Fc). The plasmid pDC316-hTERT-E1A-IRES-E1B-mCD47nb-Fc was co-transfected with the adenoviral backbone plasmid into 293 A cells to generate the recombinant virus. Viral amplification was subsequently carried out in HEK293 cells, and viral particles were purified by cesium chloride (CsCl) gradient centrifugation followed by dialysis with a specialized buffer. The final adenoviral titer was quantified using the 50% tissue culture infectious dose (TCID₅₀) method.
Ad-ON and Ad-CAM were administered to infect BMDMs at a multiplicity of infection (MOI) of 50 to generate control macrophages (Con-Ms) and CAR-Ms, respectively. The MOI is defined as the ratio of plaque-forming units (PFU) to target cells.
In vitro phagocytosis assay
The phagocytosis assay was performed in ultra-low attachment 24-well plates (Corning, 3473) to minimize tumor cell adherence. Briefly, DiD (a cell membrane fluorescent dye provided by Abkine)-labeled tumor cells (5 × 10⁴/well) were co-cultured with CAR-M cells (effector-to-target ratio = 2:1) for 5 h in RPMI-1640 supplemented with 10% FBS at 37 °C under 5% CO₂. To further prevent adherence, cells were gently resuspended every 30 min during the 4-h incubation. Phagocytosis was quantified by flow cytometry (FCM) (DiD⁺F4/80⁺ cells) and normalized to control wells (CAR-M alone).
Similarly, (CFDA-SE) (5 (6)-carboxydiacetate fluorescein succinimidyl ester) (CFSE)-labeled BMDM cells were incubated with DiD-labeled CAR-M (effector-to-target ratio = 1:1) for 5 h in RPMI-1640 supplemented with 10% FBS at 37 °C under 5% CO₂. Phagocytosis was quantified by FCM (the percent of DiD⁺ cells under CSFE gate) and normalized to control wells.
Co-incubation experiments with M2-type macrophages
On the fifth day of BMDM culture, IL-4 (20 ng/mL) was added to the medium and incubated for 48 h to induce M2 polarization. After induction, M2 macrophages were harvested and labeled with CFSE (5 µM) for 20 min at 37 °C. Following the staining process, the M2 macrophages were co-cultured with Con-M or CAR-M for 48 h. Flow cytometry was performed using anti-iNOS and anti-Arg1 antibodies, and the proportion of Arg1⁺/iNOS⁻ cells was measured to quantify the M2 macrophage population.
CAR-Ms migration assays
Migration assays for CAR-Ms were performed using 24-well plates equipped with 5.0 μm polycarbonate membrane inserts (LABSELECT, 14331-D). Briefly, 5 × 104 CAR-Ms were placed in the upper chambers containing 200 µL of RPMI 1640 supplemented with 2% FBS. The CAR-Ms were then co-cultured with 1 × 105 tumor cells (infected with oAd-CD47 or left uninfected). After 3 days, the cells remaining in the upper chamber were removed using cotton swabs, and the membrane of the insert was stained with crystal violet and imaged. The number of CAR-Ms that migrated through the membrane was quantified by counting the stained cells.
RNA sequencing
Extraction of RNA from the Con-M and CAR-M samples was conducted and a Bioanalyzer used to assess the integrity of extracted RNA. Subsequently, RNA sequencing was done by the sequencing and microarray facility, located at the Institute of Life Sciences, using an Illumina sequencer. HISAT2 RNA-seq alignment software was used to align raw sequencing reads to the mm10 reference genome (build mm10). The Feature Counts tool in Subread software was used to quantify gene expression levels. Differential expression analysis of the count data was performed using R software package DESeq2, applying a threshold of |log2(Fold Change)| ≥1 and padj ≤ 0.05 to identify differentially expressed genes. ClusterProfiler software was used to perform functional enrichment analysis for gene ontology (GO), and KEGG pathway enrichment analysis of the differentially expressed gene sets.
Real-time PCR
RNA was extracted using TaKaRaMiniBEST Universal RNA Extraction Kit (TaKaRa) and then reverse transcribed using PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa). SYBR Green III (Vazyme) was used to perform RT-qPCR. The primer sequences were listed as follows:
Spp1: AGCAAGAAACTCTTCCAAGCAA, GTGAGATTCGTCAGATTCATCCG Mrc1: CTCTGTTCAGCTATTGGACGC, CGGAATTTCTGGGATTCAGCTTC Nos2: GTTCTCAGCCCAACAATACAAGA, GTGGACGGGTCGATGTCAC Cxcl10: CCAAGTGCTGCCGTCATTTTC, GGCTCGCAGGGATGATTTCAA.
Cd86: TGTTTCCGTGGAGACGCAAG, TTGAGCCTTTGTAAATGGGCA.
Cd74: AGTGCGACGAGAACGGTAAC, CGTTGGGGAACACACACCA.
Mertk: CAGGGCCTTTACCAGGGAGA, TGTGTGCTGGATGTGATCTTC.
Fcgr1: AGGTTCCTCAATGCCAAGTGA, GCGACCTCCGAATCTGAAGA.
Animal studies
Six-week-old C57BL/6J or Balb/c female mice were obtained from Beijing Huafukang Bioscience (Beijing, China). All animal experiments were performed following the guidelines by the Animal Care and Use Committee of West China Hospital, Sichuan University, China.
In the B16 and CT26 subcutaneous tumor models, B16 or CT26 tumor cells (1 × 106 cells suspended in 100 µL DMEM) were injected into the right flank of the mice. Once the tumor volume reached about 100 mm3, the mice were randomized. CAR-Ms or Con-Ms (2 × 106cells suspended in 200 µL DMEM) were injected into the tail vein three times, with a three-day interval between each injection. The mice were intratumorally injected with the oAd-CD47 virus at a dose of 2 × 108 PFU. The length and width of the tumor were measured every two days using vernier caliper and the tumor volume calculated using the formula: tumor volume = length×(width)2 × 0.52.
For the ID8 peritoneal metastasis model, ID8-Luc tumor cells (8 × 106 cells suspended in 500 µL DMEM) were injected intraperitoneally into mice. After confirming tumor formation, mice were randomly assigned and injected intraperitoneally with CAR-Ms (4 × 106 cells suspended in 500 µL DMEM) and the oAd-CD47 virus with a dose of 0.8 × 108 PFU. Tumor progression was monitored in real time using an IVS50 bioluminescence imaging system (PerkinElmer) and analyzed with Live Image 2.6 software (PerkinElmer).
Biodistribution evaluation of CAR-Ms
The biodistribution evaluation of CAR-Ms was conducted on CT26 subcutaneous tumor model mice. The macrophages used for injection were labeled with a cell membrane near-infrared fluorescent probe (DIR). Three days post the final injection, the tumor tissues were excised and prepared for ex vivo imaging.
In vivo phagocytosis assay
Mice bearing CT26-mCherry subcutaneous tumor were treated as described in Sect. 2.9 above. Three days after the final injection, tumor tissues were excised and prepared into single-cell suspensions. Anti-CD45, anti-CD11b and anti-F4/80 was used for staining the cells and analysis performed using FCM. The percentage of mCherry+ F4/80+ cells was calculated as percentage of phagocytic activity. Similarly, ascites was collected from mice implanted with ID8-EGFP-OVA cell line as described in Sect. 2.8 and then single-cell suspensions were prepared after lysing erythrocytes. The anti-F4/80 was used to stain the cells and analysis performed using FCM. The percentage of EGFP+ F4/80+ cells was calculated to indicate phagocytosis. To investigate the role of CAR-M in the TME, the dosing regimen outlined in Sect. 2.8 was followed. DiR, a fat-soluble near-infrared fluorescent dye, was used to label CAR-M for therapy. Following the collection of a single-cell suspension, cells were stained with anti-F4/80 and anti-CD206 antibodies. The proportion of CD206⁺ DiR⁺ cells was used to identify M2-type macrophages, while the proportion of EGFP⁺ cells within the F4/80⁺ gate, further filtered by the DiR⁺ gate, was used to measure phagocytosis.
Flow cytometry analysis
The following staining protocol was used to assess CAR-PD-L1 expression on CAR-Ms: after 10-min Fc-blocking using BD Biosciences, cells were stained with Recombinant Mouse PD-L1-his (Novoprotein) and FITC anti-mouse F4/80 antibody, followed by His-tag Mouse mAb (ZEN BIO) and Rabbit Anti-mouse IgM/PE-Cy7. The following panel was used to assess the phenotypic characteristics of NC-M and CAR-Ms: anti-F4/80 FITC, anti-Arg1 PE-Cy7), and anti-iNOS APC. Additionally, the following panel was used to assess the functional analysis of NC-M and CAR-Ms: anti-F4/80 FITC, anti-MHCII PE, and anti-CD86 APC. Anti-CD47 PE and Anti-PD-L1 APC were used for CT26 and B16 tumor cells surface staining.
For analysis of the tumor and spleen microenvironment, tumor and spleen tissues were collected from mice with CT26 subcutaneous tumor model three days after the final treatment. To prepare single-cell suspensions from tumor tissue, approximately 50 mg of the resected tumor was cut into 1–2 mm³ fragments using sterile surgical scissors. The tissue was then digested in RPMI-1640 medium containing collagenase IV (1 mg/mL; Sigma, C5138) and DNase I (20 µg/mL; Roche, 10104159001) at 37 °C for 45 min with gentle agitation (150 rpm). Following enzymatic digestion, the suspension was filtered through a 70 μm cell strainer (Corning, 352350) to remove residual undigested material. Similarly, half of the spleen tissues harvested were ground with a syringe plunger and filtered through a 70 μm strainer followed by red blood cells lyses to produce a single cell suspension. The cells were then blocked with CD16/CD32 antibody and dead cells were excluded using Zombie Violet. Cell surface staining was performed at 4 °C in PBS in the dark for 25 min. For intracellular staining, the cells were fixed and permeabilized with BD Cytofix/Cytoperm Plus Fixation/Permeabilization Kit (BD Biosciences). After washing twice, the cells were resuspended in PBS for antibody staining. Flow cytometry analysis was performed using a BD Biosciences LSRFortessa, and the data analyzed using flowjo software (Tree Star Inc.). The following antibodies were used in cell staining:
Panel 1 was used for T cells staining in tumor: CD3-Texas Red, CD4-BUV395, CD8-PE-Cy7, PD-1-Percp, Tim3-PE, CD25-FITC, and Foxp3-APC.
Panel 2 was used for TAMs/dendritic cells (DCs)/MDSCs: CD45-APC-cy7, CD11b-APC-cy5, F4/80-BUV730, CD206-PE-cy7, MHCII-BV711, GR1-BV786, CD86-APC, CD11c-BV650, and CD80-FITC.
Panel 3 was used for T cells staining in spleen: CD3-Texas Red, CD4-FITC, CD8-BV510, TNF-α-APC, and IFN-γ-PE.
All antibodies for flow cytometric analysis were obtained from Biolegend. Samples for assessing IFN-γ and tumor necrosis factor-alpha (TNFα) were stimulated with phorbol 12-myristate 13-acetic acid (PMA) and ionomycin for 2 h before fixation and staining.
OVA specific-T cell response assay
ID8-EGFP-OVA tumor cells (8 × 106 cells suspended in 500 µL DMEM) were injected intraperitoneally into mice. Seven days after the final treatment, spleens were aseptically excised and lymphocyte suspensions were harvested. Lymphocytes were plated at a density of 1 × 105 cells per well in anti-IFN-γ precoated plate and incubated with OVA peptide (final concentration: 10 µg/ml) for 24 h at 37 °C. IFN-γ production was assessed using Mouse IFN-γ ELISpot kit and visualized using ImmunoSpot S6 Universal. Quantification of IFN-γ was performed using ImmunoSpot V.7.0.15.0 Professional Analyzer DC software.
Neoantigen-specific T cell response assays
Spleens were prepared as discussed in 2.12 above and seven days after treatment, spleen were aseptically excised and prepared and lymphocyte suspensions harvested. Lymphocytes were plated at a density of 1 × 105 per well in 96-well plates and incubated for three days with OVA peptide (final concentration: 10 µg/ml), 6 neoantigen peptides, and mixed peptide pools (final concentration of each antigen peptide: 2 µg/ml) in the presence of complete medium supplemented with 100 µg/ml double antibodies, 50 µM 2-mercaptoethanol, and 300 IU/ml recombinant mouse IL-2, and 100 µL supernatant was collected from each well and IFN-γ expression levels assessed using Mouse IFN-γ Precoated ELISA kit (YOTA. The remaining cells were used for Edu-488 cell proliferation assay kit (Beyoclick) to assess T cell proliferation after neoantigen stimulation. CT26 neoantigen peptides were synthesized using Fmoc-based solid-phase synthesis (GL Biochem, Shanghai), incorporating N-terminal acetylation and C-terminal amidation. The synthesized peptides were purified to > 95% purity via reversed-phase HPLC), and their identities were verified by MALDI-TOF mass spectrometry. The sequence of the neoantigen peptide is presented as follows [27]: 1: Aldh18a1: HSGQNHLKEMAISVLEARACAAAGQ; 2: E2f8: ILPQAPSGPSYATYLQPAQAQMLTP; 3: Ndc1: HSFIHAAMGMAVTWCAAIMTKGQYS; 4: Slc20a1: KPLRRNNSYTSYIMAICGMPLDSFR; 5: Mtch1: SWIHCWKYLSVQSSQLFRGSSLLFRR; 6: Dhx35: VIQTSKYYMRDVIAIESAWLLELAP.
In vivo T cell depletion experiments
Anti-mouse CD4-Invivo (Selleck) and anti-mouse CD8-Invivo (Selleck) were injected intraperitoneally at a dose of 200 µg per mouse every three days, for a total of three days, starting at two days before treatment initiation. Three days after the first injection, peripheral blood monocyte suspension was obtained and stained with anti-mouse CD3, CD4, and CD8 antibody to assess the blocking efficacy. Tumor assessment procedures were conducted as described in Sect. 2.13.
Statistical analysis
Statistical analyses were performed using Graphpad prism software version 8.0 and IBM SPSS software version 27. A two tailed Student’s t-test was used for comparison between the two groups, whereas one-way analysis of variance (ANOVA) was applied for multiple group comparisons. All experiments were independently repeated [n] times with consistent results, where [n] represents the biological replicates. The figure presents representative data from at least three independent experiments (n ≥ 3), with the data expressed as the mean ± the standard error of the mean (SEM). Significance levels were denoted as, *p < 0.05, **p < 0.01, ***p < 0.001, and N.S for not significant.



Comments (0)