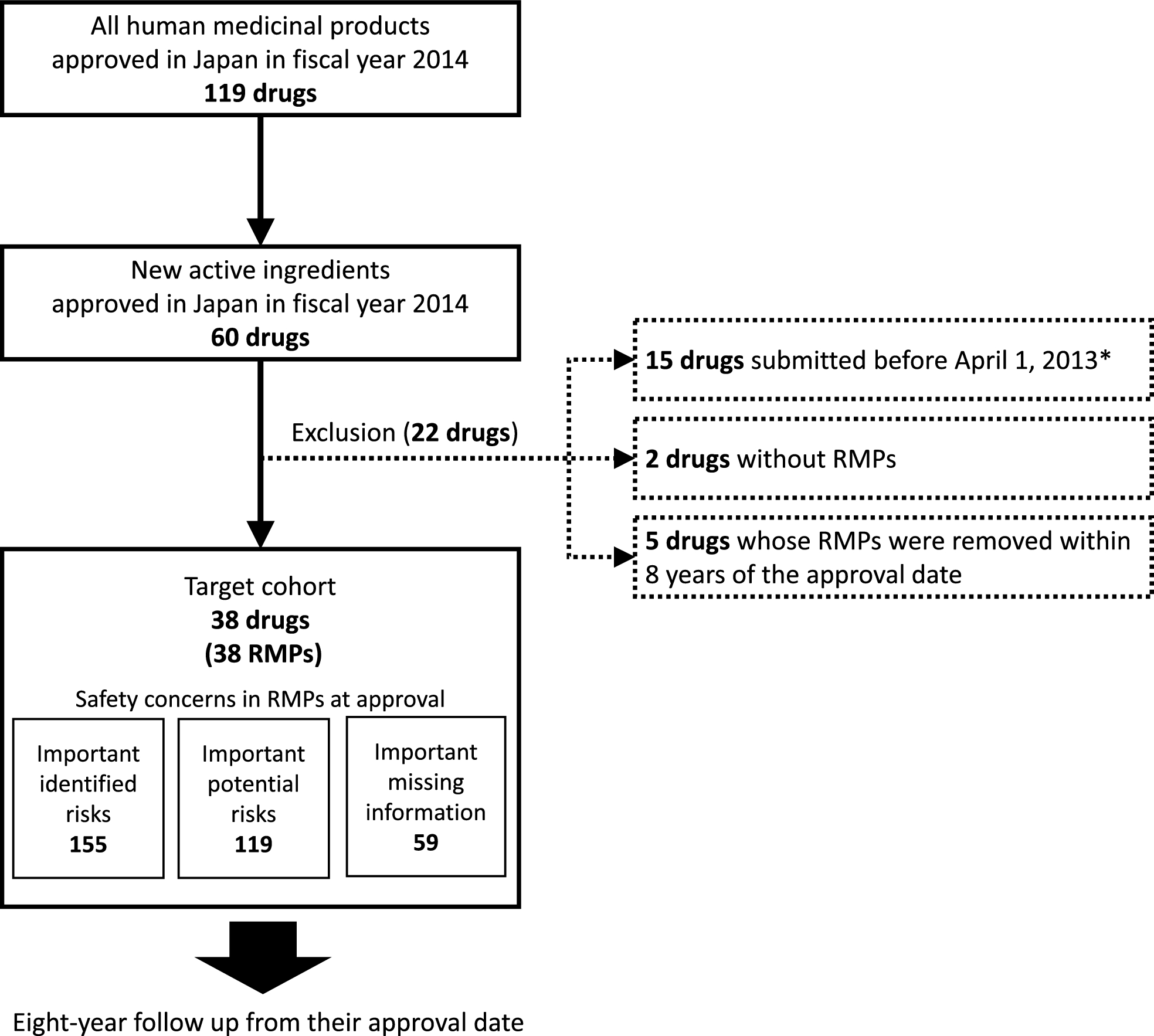
Our longitudinal analysis of RMP safety concerns for 38 new drugs approved in 2014 identified 155 IIRs, 119 IPRs, and 59 IMIs in the first edition. After 8 years, most IIRs and over 88% of the IPRs and IMIs remained; 29 IIRs, 20 IPRs, and 3 IMIs were added; 14 IPRs were upgraded to IIRs; and 7 IMIs were removed. More dynamic changes were especially noted for products with additional approvals, but most of the evidence for these changes was based on pharmacovigilance data rather than on clinical trial data.
These results suggested that pharmacovigilance activities occupy a vital role in safety assessment in the post-marketing phase in Japan. First, the number of IIRs added after approval (where no signal for such risks was found in pre-approval clinical trials) accounted for about 15% (29/198) of all IIRs. Second, the changes of newly added IIRs or IPRs and IPRs upgraded to IIRs were based on evidence gathered through pharmacovigilance activities rather than from clinical trials. Third, all new IMIs added during the follow-up period were added due to the results of clinical trials conducted for approvals of additional indications. Lastly, 7 IMIs were removed from RMPs during follow-up based on pharmacovigilance data. All these results not only highlight the importance of pharmacovigilance activities but also the limitations of clinical trial conclusions for use in safety assessments. It is known that there are five limitations to clinical trials from the perspective of safety evaluation: too few (small number of patients), too simple (simple design), too brief (short administration period), too median-aged (no children or elderly patients), and too narrow (narrow range of indications) [13]. Our results show that these limitations of clinical trials were complemented by pharmacovigilance activities.
All IIRs in RMPs at first approval remained for 8 years. There is a definition for the inclusion of IIRs in RMPs, but no definition for the removal of IIRs from RMPs in the Japanese RMP guideline [1]. Meanwhile, the EU RMP guideline, Guideline on good pharmacovigilance practices Module V, allows for the deletion of IIRs under certain circumstances; for example, when the risk is fully characterized and appropriately managed, IIRs may be removed from the safety specification [14]. The content of this guideline was revised in 2017; since then, the number of safety concerns in existing EU RMPs decreased [9]. According to the EU guidelines, IIRs can be removed if the risk is adequately characterized by pharmacovigilance activities and adequately managed by risk minimization activities. It is unclear whether IIRs remained in Japanese RMPs because pharmacovigilance and risk minimization activities have not been functional, or because they have been functional but have not warranted the removal during the re-examination period. In either case, the current situation is problematic; in the former case, sufficient evaluations have not been carried out and risks have not been minimized; in the latter case, RMPs were not updated regularly and did not focus on important aspects of patient safety in clinical practice based on the latest information. Therefore, it is imperative that appropriate pharmacovigilance and risk minimization activities are planned, implemented, and evaluated, and that the need for IIRs is reconsidered based on the latest information.
Of the IPRs present at first approval, 88% remained as IPRs, 12% were changed to IIRs, and none were removed from the RMPs during the 8 year study period. IPRs are those safety concerns for which a causal relationship could not be established at the time of approval owing to a lack of evidence, and for which further information continues to be gathered through routine and additional pharmacovigilance activities. In this study, half of the target drugs were non-orphan drugs and had completed the re-examination period at the end of the follow-up period; i.e., even though all pharmacovigilance activities necessary to address safety concerns had been completed, most IPRs remained. These results could indicate that additional or routine pharmacovigilance activities planned at the time of approval were insufficient to assess causality, or that these activities did not show causality but the IPRs remained because there are no criteria for their removal from the RMPs. The former issue may be due to the fact that post-marketing studies without a control group through primary data collection account for about 80% of all additional pharmacovigilance activities in Japan [15]. Because potential risks identified in clinical trials often require quantitative analysis with a control group to assess causality in the post-marketing setting, causality is challenging to assess based on the results of such studies without a control group. Recently, the number of health information databases available for epidemiological studies in Japan has been increasing [16]. It is necessary to plan additional and more appropriate pharmacovigilance activities using these databases to generate evidence. On the other hand, regarding the latter issue, similar to the IIR issue, a standard procedure for deletion may be needed.
Subgroup analyses showed a trend toward more changes in IIRs, IPRs, and IMIs in the group with additional approvals. However, only a limited number of changes were based on clinical trials conducted for additional approvals. These results may be because drugs with additional approvals have more opportunities to revise their RMPs and because more safety information is available from safety pharmacovigilance activities since the additional indications increase the number of patients using these drugs.
There are several limitations to this study. First, only drugs with new active ingredients approved in 2014 (immediately after the introduction of RMPs in Japan) were included because we needed at least 8 years of follow-up. However, we believe that the results of this study could be extrapolated to drugs approved in other years because the procedure for IIRs, IPRs, and IMIs has currently not changed, and because target drugs with various drug classes were included. Second, the 8-year follow-up period meant that orphan drugs could not be followed up to the end of their re-examination period (10 years). In addition, we were unable to obtain the results of PMDA re-examination reviews for most drugs. Marketing authorization holders generally must submit a re-examination application within 3 months of the end of the re-examination period and the PMDA re-examination review process takes additional time (usually more than one year) from submission. Therefore, further long-term research is needed to comprehensively assess the impact of the re-examination review process on safety concerns in RMPs. Third, the details of the information supporting RMP changes could not be determined because the reasons for RMP changes were not public. Further research and public information on the drivers of RMP changes are needed to improve the functioning of the J-RMP.


Comments (0)