
Remember me
We here report the final results of the multicenter prospective randomized phase II PORTAF trial (ClinicalTrials.gov: NCT02189967) investigating whether accelerated PORT (7 fractions per week, 2 Gy per fraction) improved LRTC compared to conventional fractionation (5 fractions per week, 2 Gy per fraction) in patients with NSCLC. We used the CONSORT reporting guidelines.
Recruitment, randomization, and workflowThe study protocol and associated procedures have been published previously [14]. In brief, all patients with signed informed consent were included in the study. Before randomization blood analyses and postoperative pulmonary function test needed to be done as well as a complete staging including FDG-PET/CT or, alternatively, contrast-enhanced CT of the chest/abdomen and bone scintigraphy and cerebral MRI in cases of suspected brain metastases. For patients receiving postoperative adjuvant chemotherapy, these restaging procedures had to be repeated prior to the start of radiotherapy.
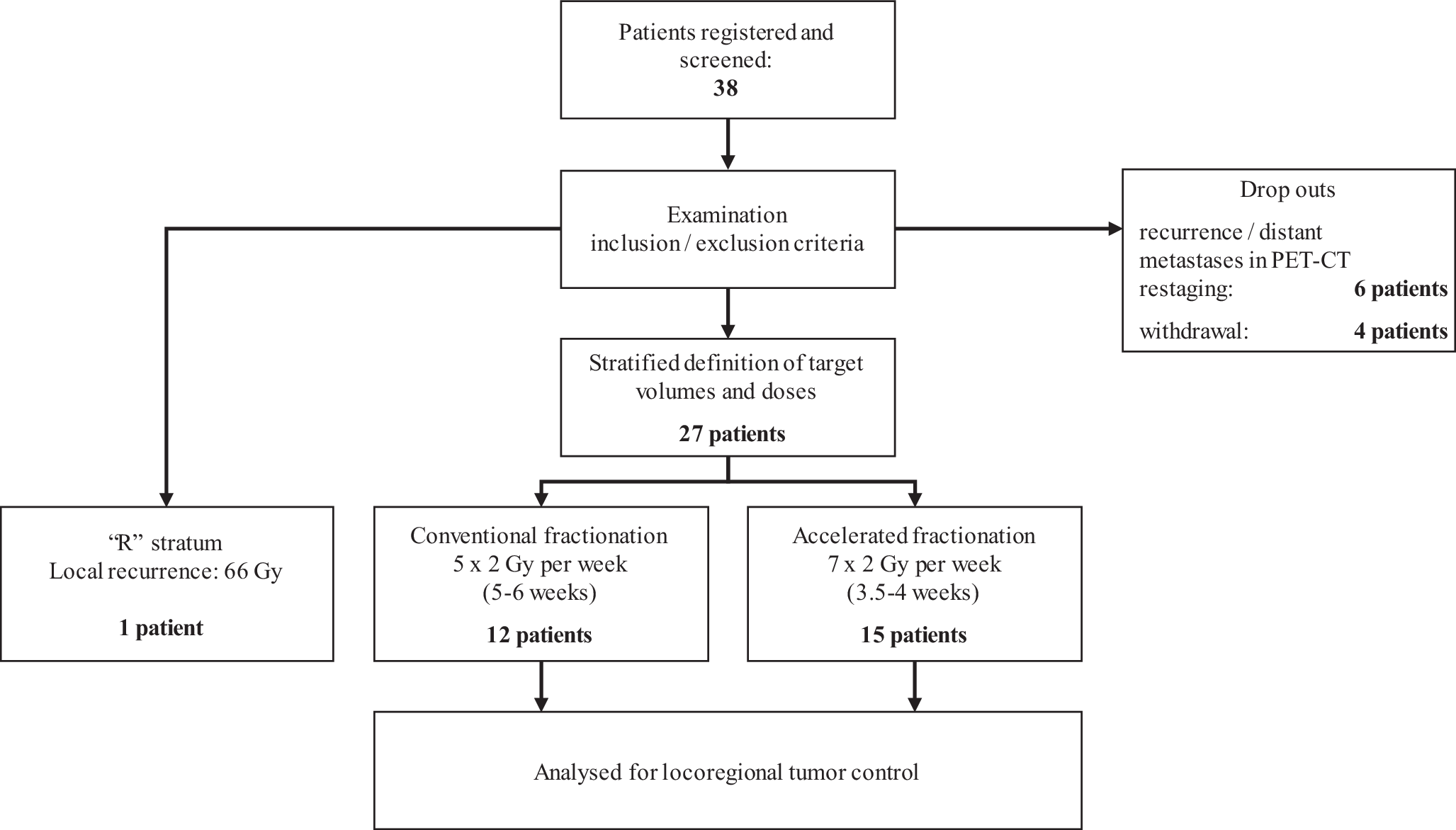
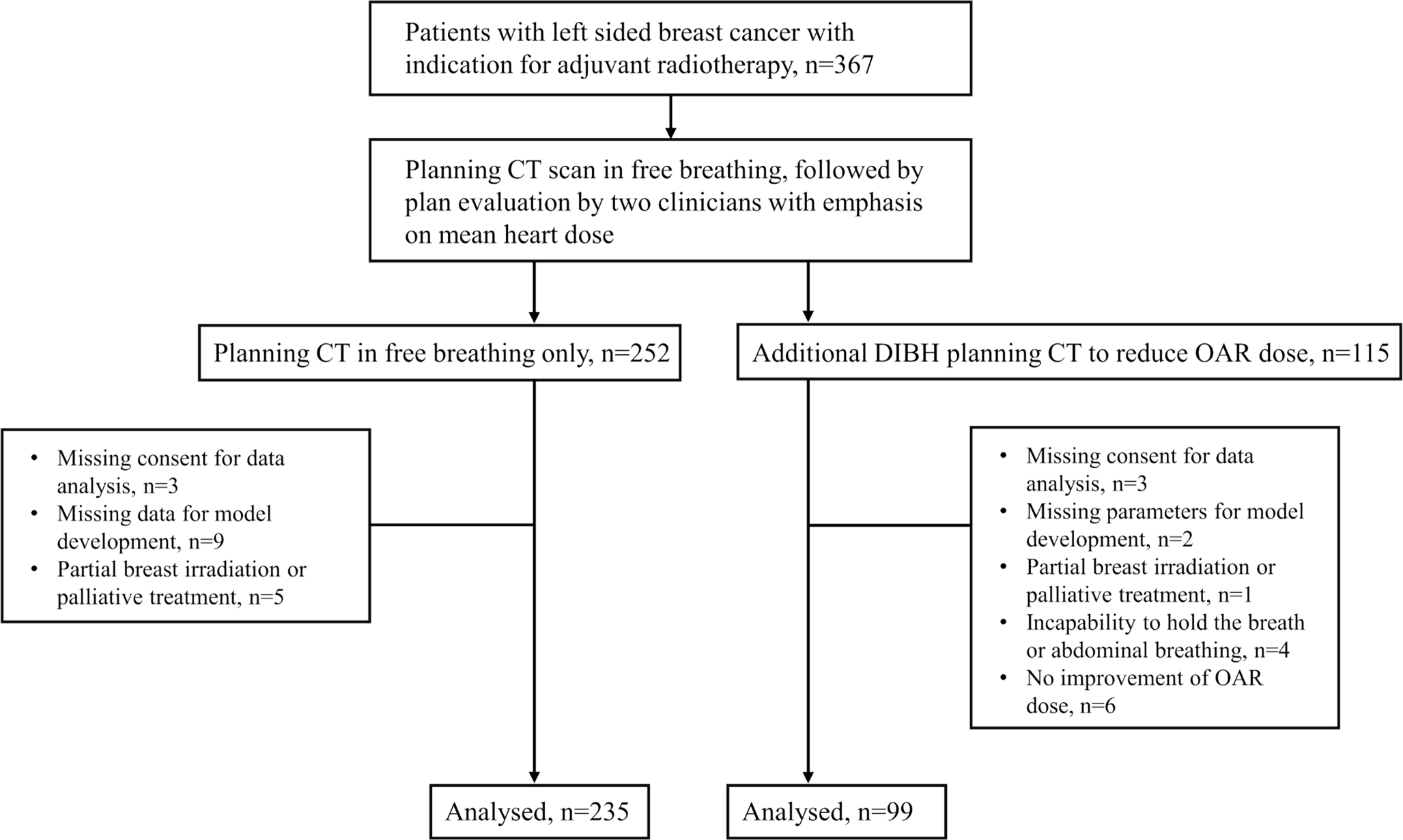
For electronic data collection and randomization, the RadPlanBio platform [15] was used by each study site, with stratification of patients by the following criteria: staging with or without PET/CT, resection status R1 or R0, and the respective study center. Patients with macroscopic tumor detected in pre-irradiation staging were treated in stratum “R” of the trial and received conventionally fractionated radiation therapy to a total dose of 66 Gy. Figure 1 shows a flowchart of the study and Table 1 the inclusion and exclusion criteria.
Fig. 1
Flowchart of the study procedures
Table 1 Inclusion and exclusion criteriaPrimary and secondary endpointsThe primary endpoint of this study was locoregional tumor control 36 months after the start of radiotherapy comparing an accelerated (7 fractions per week, 2‑Gy single dose) with the standard conventionally fractionated schedule (5 fractions per week, 2‑Gy single dose) for postoperative radiotherapy in patients with NSCLC.
Locoregional tumor control was defined as freedom from local recurrence and regional recurrence (in-field, at the edge of the field, or out of field). Secondary endpoints included overall survival, local recurrence-free and distant metastases-free survival 36 months after the start of radiotherapy, acute and late toxicity, and quality of life for both treatment methods.
Clinical follow-up examinations with imaging scans (FDG-PET/CT or CT of the thorax/abdomen) were scheduled every 3 months within the first 3 years and every 6 months for the following 2 years. During radiation therapy and at the follow-up visits, scoring of side effects was performed according to the Common Terminology Criteria for Adverse Events (CTC-AE) 4.0. Quality of life was assessed additionally by the European Organisation For Reserach and Treatment of Cancer (EORTC) questionnaires QLQ-C30 and QLQ-LC13 before and at the end of irradiation and at each follow-up visit.
The trial design and protocol adhere to the SPIRIT criteria (Standard Protocol Items: Recommendations for Interventional Trials; www.spirit-statement.org) [14].
Biometric designTarget volumes and total radiation doses were stratified in both treatment arms based on individual risk factors. The initial sample size calculation was based on the hypothesis of an improvement of 15% in the primary endpoint, i.e., an increase in the locoregional tumor control rate from 70 to 85% after 3 years in the accelerated treatment arm (7 fractions per week) in the group of R0- or R1-resected patients. Assuming α to be 0.05, a rise of 15% could be detected with a power of 80% if 154 patients were to be treated in each arm. This number includes a dropout rate of 10%.
The observational “R” stratum, which contains patients with macroscopic remaining tumor or recurrences detected in pre-irradiation staging, was not included in the statistical sample size calculation.
RadiotherapyFor radiation planning purposes, all patients should preferably have undergone FDG-PET/CT in the radiation treatment position. On the basis of the planning CT, the clinical target volume and the organs at risk were contoured in accordance with the trial protocol. Standardization of contouring was achieved by a mandatory dummy run for all participating centers with quality checks by three experienced radiation oncologists (SA, RB, ET). Each patient was treated with an image-guided conformal radiation technique (3D-CRT; intensity-modulated RT, IMRT).
The mediastinal target volume received a total dose of 50 Gy, and in the case of microscopic incomplete resection (R1) or extracapsular tumor spread (ECE), additional boost irradiation of 10 Gy (2 Gy per fraction) to a total dose of 60 Gy was delivered. Accelerated radiotherapy was conducted by application of a second fraction on two weekdays and/or an additional irradiation at weekends. It was mandatory to have an interval of at least 6 h between treatment fractions, preferably 8 h.
Data monitoring committeeThe Data Monitoring Committee (DMC) consisted of five members: four clinicians with experience in the study-specific indication area of radiotherapy (V. Budach, R. D. Kortmann, R. Schwarz, D. Vordermark) and one biometrician (P. Martus). The members of the DMC were independent from the study centers and have declared that there exists no scientific or financial conflict of interest. The DMC reviewed the results of safety analyses and quality controls as well as the evaluations of toxicities and their impact on the benefit–risk assessment for the patients twice.
Comments (0)