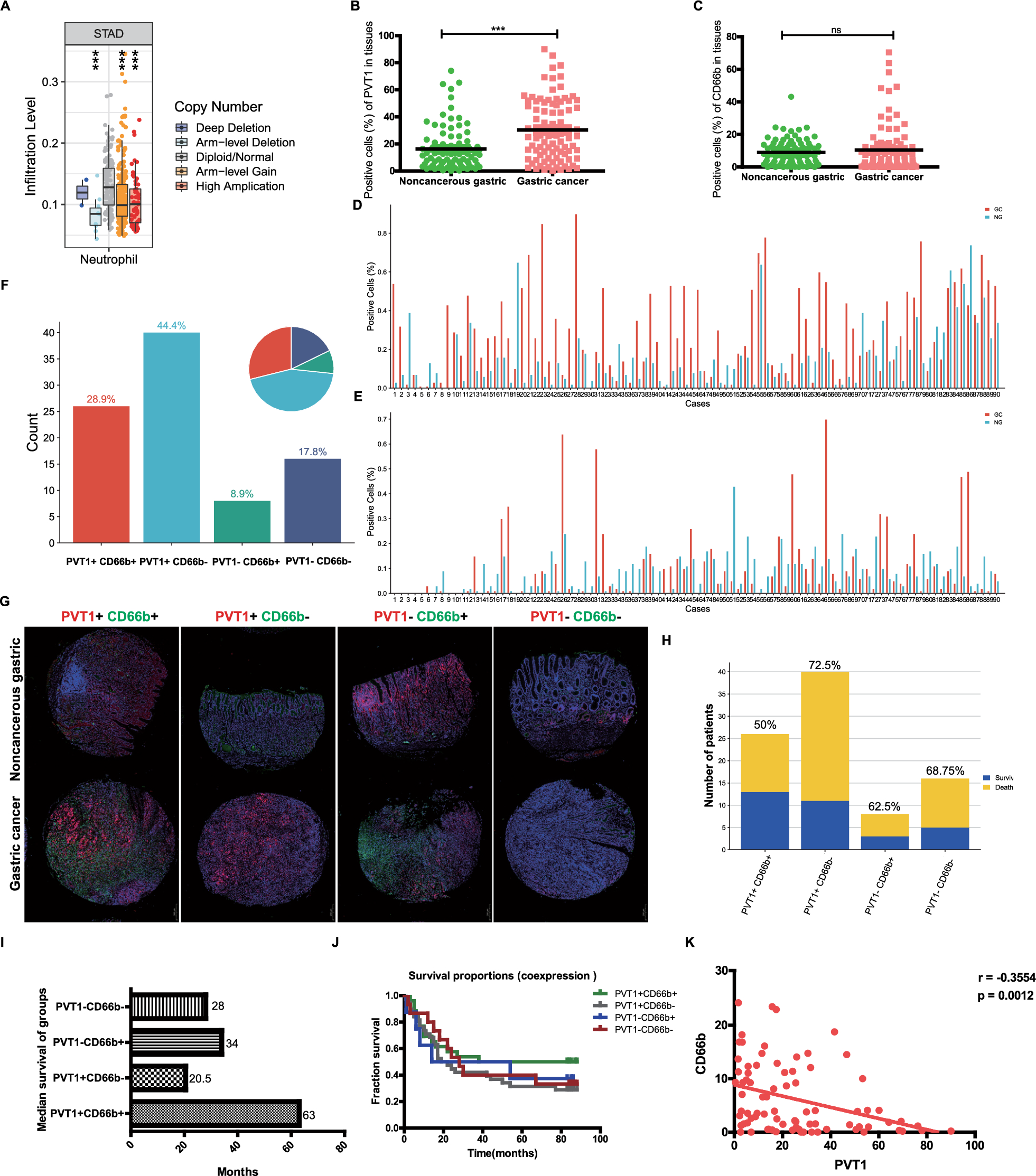
Gastric cancer tissues chip of patients, blood in healthy population
The tissue chip consisted of 90 cases of human gastric cancer tissues and their corresponding adjacent tissues, totaling 180 sites (number: HStmA180Su11, lot number: XT17-035), obtained from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). The chip was accompanied by matching HE staining information. Among the samples, there were 32 patient fatalities and 58 survivors. The peripheral blood samples were obtained from 10 healthy subjects at the Second Affiliated Hospital of Harbin Medical University following routine examination, collected in EDTA-K3 anticoagulant tubes. This study received ethical approval from the Medical Ethics Committee of the Second Affiliated Hospital of Harbin Medical University prior to commencement (Approval number: YJSKY-2022-517).
Cell culture, transfection and treatment
The human gastric mucosal epithelial cell line GES-1 and the human gastric cancer cell lines HGC-27, AGS, NCI-N87 and MKN-45 were maintained in accordance with a previous report [26]. GC cell line SNU-16 was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 (Gibco BRL, Waltham, MA, USA) with 10% fetal bovine serum at 5% CO2 in a 37 ℃ incubator. The genotypically validated of cell lines were authenticated by Beijing Microread Genetics Co., Ltd. (Beijing, China) using short tandem repeat (16-STR) analysis. The antisense oligonucleotide (ASO) for PVT1 and negative control (NC) were purchased from Guangzhou RiboBio Co., Ltd. (lnc6180719010843-1-5, Guangzhou, China). Cells were seeded in six-well plates, and transfected with PVT1-ASO or NC using riboFECTTMCP Transfection kit (C10511-1, RiboBio, Guangzhou, China) according to the instructions. The transfection efficiency was measured by real-time PCR at 48 h and 72 h after transfection. For tumorigenicity assay in vivo, lentiviral particles for PVT1-shRNA(Catalog #: LPP-CS-SH3617-LVRU6GP-a-500) and for negative control (Catalog #: LPP-CSHCTR001-LVRU6GP-500) were purchased and infected to HGC-27 cells using the Lentiviral granulosa Cell Infection Experiment User's Manual from GeneCopoeia, Inc. Recombinant Human TNF-α was purchased from Beyotime Biotechnology (p5318, Shanghai, China) and recombinant Human SYNPO protein was purchased from Zeye Biotechnology (ZY685Hu01P, Shanghai, China).
Isolation, extraction, and identification of neutrophils from healthy individuals
The isolation of peripheral blood neutrophils was conducted following the protocol from the human peripheral blood neutrophil isolation kit (Solarbio, P9040, Beijing, China). Briefly, 4 mL of reagent A was layered with 2 mL of reagent C in a 15 mL tube, followed by overlaying 5 mL of anticoagulated blood. The mixture was centrifuged at 2500 rpm for 30 min at room temperature to separate the mononuclear cell layer (upper) and neutrophil layer (lower). Neutrophils were aspirated, washed with 10 mL washing solution (1200 rpm for 10 min), lysed with 2 mL lysis buffer for 10 min, neutralized with 5 mL washing solution, centrifuged, and resuspended in culture medium.
After isolation, cell viability was assessed using the Trypan Blue staining kit (Beyotime, C0011, Shanghai, China) and an automated cell counter (LUNA-II™, Logos Biosystems, Shanghai, China). Neutrophil purity was determined by Wright’s stain solution (Solarbio, G1040, Beijing, China), followed by observation and photography under an optical microscope (ZEISS Axio Vert.A1, Germany).
Dual hybridization of fluorescence in situ hybridization (FISH) and Immunofluorescence (IF)
To account for inter-individual variability in GC patients, the percentage of positive cells was used to evaluate the expression levels of PVT1 and neutrophil infiltration in both tumor tissues and adjacent tissues obtained from the same GC patient. The tissues were deparaffinized and subjected to antigen retrieval according to standard protocols. Subsequently, the samples were digested with proteinase K, pre-hybridized, and hybridized with a PVT1 fluorescent probe (1:67 dilution, Genepharma) combined with the RNA FISH SA-Biotin amplification system kit (Genepharma). The tissues area was sealed at room temperature, followed by the addition of the anti-CD66b primary antibody (1:500, Abcam, ab300122). After overnight hybridization at 4 ℃, incubation with FITC-labeled goat anti-rabbit secondary antibody (1:500, Immunoway, RS0004) was performed. Finally, the DAPI dye solution was added. The tissues were examined using a fluorescence microscope (NIKON, YS100, Japan) and images were acquired. CD66b-positive cells emitted green fluorescence, PVT1 produced red fluorescence, and DAPI-stained nuclei showed blue fluorescence.
Whole transcriptome sequencing (WTS)
WTS was conducted by Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). Briefly, the total RNA was extracted using the RNeasy Mini Kit (Qiagen, 74106, Germany). Sequencing libraries were generated using VAHTS™ Stranded mRNA-seq Library Prep Kit for Illumina (Vazyme, NR612, Nanjing, China) according to the manufacturer’s instructions. The sequencing was performed on Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). Raw data were processed through Seqtk. In this step, the raw data were cleaned by removing adapter sequences, contaminant reads, and low-quality bases. Additionally, the Q30, and GC content were calculated to estimate the quality of clean reads. Next, the clean reads were aligned to the reference genome (GRCh38) using HISAT2. The Read Counts of transcripts and ncRNAs were calculated by String Tie. And the expression of mRNA and lncRNA was normalized to FPKM. FPKM refers to the number of fragments per kilobase length from a gene per million fragments mapped considering the effect of both sequencing depth and gene length on fragments count. The sequencing depth (in total reads) of WTS is summarized in Table S1.
4D-Label-free quantitative proteome analysis by LC–MS/MS
LC–MS/MS was conducted by Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). It comprised protein extraction and peptide hydrolysis, as well as LC–MS/MS data collection. The samples were lysed by SDT (4% (w/v) SDS, 100 mM Tris–HCl pH 7.6, 0.1 M DTT). Each sample was separated using Brucker nanoElute, a ultra performance liquid chromatography (UPLC) liquid phase system with a nanoliter flow rate. Samples separated by nanoflow ultra-high performance liquid chromatography (nanoUHPLC) were analyzed by data-dependent acquisition (DDA) mass spectrometry using timsTOF Pro mass spectrometer (Bruker). The detection mode was positive ion mode, and the ion source voltage was set to 1.5 kv. Time of flight (TOF) was used for detection and analysis of MS and MS/MS. The mass spectrum scanning range was set to 100–1700 m/z. Parallel accumulation aerial fragmentation (PASEF) mode was used for data acquisition.
Real time quantitative polymerase chain reaction (Real-time PCR)
Total RNA were extracted from cells using the SevenFast™ Total RNA Extraction Kit (SEVEN, SM130, Beijing, China), and first-strand cDNA was synthesized using the All-in-one First Strand cDNA Synthesis Kit (SEVEN, SM131, Beijing, China) according to the manufacturer’s protocol. The cDNA was subjected to real-time PCR in SLAN-96P real-time PCR system (Hongshitech, Shanghai, China) using 2 × SYBR Green qPCR Master Mix (SEVEN, SM133, Beijing, China) with fluorescence detection. Genes expression levels were normalized to β-actin by the 2−△△Ct method. The sequence of primers are listed in Table S2.
Western blot analysis
Total protein was extracted from the GC cells using RIPA buffer (Beyotime, P0013B). The protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk for 2 h at room temperature and then hybridized with primary antibodies: anti-Synaptopodin (1:1,000; Affinity, DF12173), anti-MMP9 (1:1,000; Beyotime, AF5234) and anti-β-actin (1:10,000; Affinity, AF7018) overnight at 4 °C, followed by incubation with a horseradish peroxidase HRP-conjugated goat anti-rabbit secondary antibody (1:10,000; ZSGB-BIO, ZB-5301). Blots were imaged using chemiluminescence imaging system (ChemiScope 6300, CLiNX).
CCK-8 assay and cell viability analysis
Cells were seeded into 96-well plates at a density of 2000 cells per well. Cell proliferation was monitored daily over 5 consecutive days, while viability was assessed at 16 h or 24 h using the Cell Counting Kit-8 (SEVEN, SC119, Beijing, China) according to the manufacturer’s protocol. Optical density (OD) was measured at 450 nm using a microplate reader (BIO-RAD, iMark, Japan). Each experiment included six biological replicates, and data are expressed as mean ± standard deviation (SD).
$$\text(\text) =\frac\times 100$$
Colony formation assay
Cells were seeded in 6-well plates at a density of 200 cells per well. Following 10 days of culture, the medium was removed, and cells were washed twice with phosphate-buffered saline (PBS). Subsequently, cells were fixed with methanol for 20 min at room temperature, followed by PBS washing and staining with Giemsa solution (Beyotime, C0133, Shanghai, China) for 30 min. The assay was performed in three independent replicates. Colonies were imaged and quantified with ImageJ software.
Wound healing assay and transwell migration assay
Cell migration ability was evaluated through wound healing and transwell assays.
Wound healing assay
Cells were seeded in 6-well plates and cultured until reaching 90% confluency. A uniform scratch was created using a sterile 200 μL pipette tip. After washing with PBS to remove detached cells, cells were maintained in medium containing 2% FBS (to suppress proliferation). Wound closure was monitored at 0 h and 24 h using an inverted microscope (ZEISS, Axio Vert.A1, Germany).
Transwell migration assay
Transwell chambers (8 μm pore size, Corning, NY, USA) were placed in a 24-well plate. The upper chamber was loaded with 200 μL serum-free medium containing 3 × 104 cells, while the lower chamber contained 800 μL medium with 10% FBS as a chemoattractant. After 24 h incubation, non-migrated cells on the upper membrane surface were removed with a cotton swab. Migrated cells on the lower surface were fixed with 4% methanol for 20 min, stained with Giemsa solution (Beyotime, C0133, China) for 30 min, and imaged under an optical microscope (ZEISS, Axio Vert.A1, Germany).
IF, apoptosis/necrosis, and Fluo-4 calcium assaysIF
Cells were cultured in 6-well plates. DNA damage was assessed using the DNA Damage Assay Kit (Beyotime, C2035S) according to the manufacturer’s protocol. For protein detection, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% BSA. Subsequently, cells were incubated with anti-γ-H2AX antibody (Beyotime, C2035S-4), anti-SYNPO (1:500, Affinity, DF12173), anti-SERPINE1 (1:200, Immunoway, YT3569), anti-TP53 (1:200, Immunoway, YT3528) and anti-CHEK1 (1:200, Immunoway, YT0902) overnight at 4 °C. After washing, cells were incubated with either Alexa Fluor 488-conjugated anti-rabbit IgG (Beyotime, C2035S-5) or Goat Anti Rabbit IgG (H + L) (AbFluor 594) (1:200, Immunoway, RS3611) for 1 h at room temperature. Cells were counterstained with DAPI (Beyotime, C1002) or Phalloidin (YEASEN, 40774ES03) and imaged using a fluorescence microscope (NIKON, YS100, Japan).
Apoptosis and necrosis assay
Cell apoptosis and death was analyzed using the Apoptosis and Necrosis Assay Kit (Beyotime, C1056). Briefly, cells were stained with Hoechst 33,342 (10 μg/mL) and propidium iodide (PI) (5 μg/mL) in staining buffer for 20 min at 37 °C. After PBS washing, apoptotic (Hoechst-positive) and necrotic (PI-positive) cells were quantified under a fluorescence microscope (NIKON, YS100, Japan).
Fluo-4 calcium assay
Intracellular calcium levels were measured with the Fluo-4 Calcium Assay Kit (Beyotime, S1061S). Cells were loaded with Fluo-4 AM in staining solution at 37 °C for 30 min. Green fluorescence signals (Ex/Em = 490/525 nm) were captured using a fluorescence microscope (NIKON, YS100, Japan). Data were normalized to baseline fluorescence intensity.
Tumorigenicity assay in vivo
Female BALB/c Nude mice aged 4–6 weeks were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and were housed under pathogen-free conditions. Mice were randomly divided into NC groups (n = 5) and experimental groups (n = 5). Subcutaneous injections of HGC-27 cells (1 × 107) with or without stable PVT1 silencing were administered to establish xenografts. Tumor growth was monitored for 4 weeks before mice were sacrificed. Mice were sacrificed by cervical dislocation and tumor tissues were obtained for pathological examination. Paraffin sections of tumors were prepared by conventional methods, and the tumor tissues were observed and examined via hematoxylin and eosin (HE) staining. Ki67 expression was assessed by immunohistochemistry using an anti-Ki67 antibody (1:100, Affinity, AF0198) in tumor tissues among different groups. All animal studies were performed in accordance with the ethical principles of animal protection and welfare, and approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (Ethical review approval number: YJSDW2022-252).
Statistical analysis
Data are expressed as mean ± SD and analyzed using GraphPad Prism software (v10.1.2). Comparisons between two groups were performed using the Wilcoxon rank-sum test or Student’s t-test. A p < 0.05 was considered statistically significant unless otherwise stated. Sequence data visualization was generated by https://www.bioinformatics.com.cn (last accessed on 20 Feb 2023), an online platform for data analysis and visualization.






Comments (0)