
Remember me
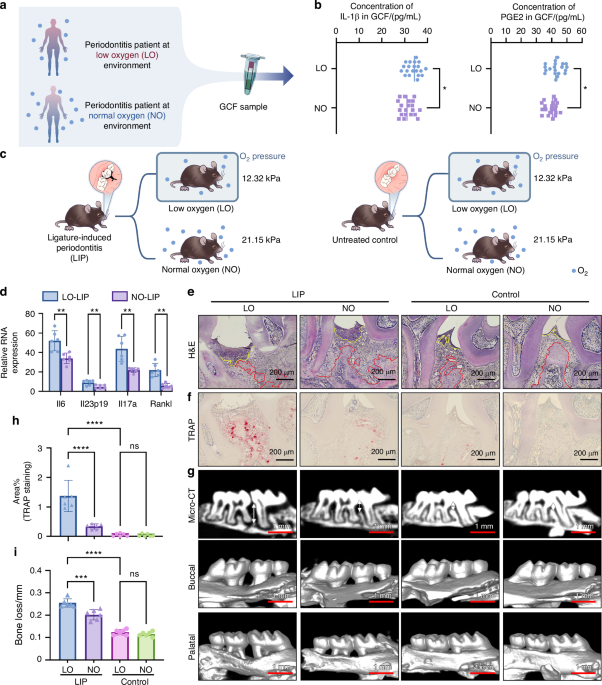
To investigate the relationship between hypoxia and oral mucosal inflammation, we first compared gingival crevicular fluid (GCF) from periodontitis patients in low-oxygen (LO; n = 20) and normal oxygen (NO; n = 20) environments. These patients were selected after carefully excluding known confounders or comorbidities. The definitions of LO and NO are detailed in the Methods section. The levels of interleukin-1β and prostaglandin E2 in GCF were measured. Results indicated significantly elevated concentrations of these inflammatory cytokines in LO patients (Fig. 1a, b).
Fig. 1
Hypoxia aggravates periodontitis in vivo. a Schematically depicting the procedure of human sample collection. b Concentration of IL-1β (left) and PGE2 (right) in the GCF of periodontitis patients in LO or NO environment (n = 20). c Schematically depicting the design of in vivo experiments. d Relative mRNA expression of inflammatory cytokines (Il6, Il23p19, Il17 and rankl) in mouse gingiva subjected to LIP, under LO or NO environment (n = 6). e Representative images of H&E staining. Scale bars, 200 μm. The red dash represents the boundary between bone and connective tissue. The yellow dash represents the boundary between connective and epithelium. f, g Representative images of TRAP staining (f) and Micro-CT (g) of the periodontal tissue of mouse under LO or NO environment, with or without subjected to LIP. Scale bars of TRAP staining, 200 μm. Scale bars of Micro-CT, 1 mm. h Semi quantitative analysis of TRAP staining in f (n = 6). i Differences in bone loss among mouse in different groups (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001; **P < 0.01; *P < 0.05; ns, no significance. Statistical significance was determined by two-tailed unpaired Student’s t test (b, d) or one-way ANOVA test (h, i). IL-1β, Interleukin-1 β, PGE2 Prostaglandin E2, GCF gingival crevicular fluid, LO low oxygen, NO normal oxygen, H&E hematoxylin and eosin, TRAP tartrate resistant acid phosphatase, LIP ligature-induced periodontitis, micro-CT micro-computed tomography)
To further examine this correlation, we established a murine ligature-induced periodontitis (LIP) model under hypobaric hypoxia (Fig. 1c). Consistent with the GCF findings, inflammatory gene expression was higher in LIP mice exposed to LO conditions (Fig. 1d). In addition, LO-exposed LIP mice exhibited increased bone destruction and more severe periodontitis, whereas LIP mice in NO conditions experienced minimal disease progression (Fig. 1e–i). These findings suggest that hypoxic environments exacerbate periodontitis. Further research is required to elucidate the precise mechanism by which hypoxia contributes to disease progression.
Since the oral microbiome plays an important role in periodontitis, we next examine whether hypoxia-induced changes in the microbiome contribute to disease exacerbation. 16S rRNA gene sequencing was used to characterize the microbiome. However, microbiome composition and structure showed no significant differences between LO-LIP and NO-LIP mice (Fig. S1a–c). Although previous studies have reported substantial microbiome shifts under hypoxic conditions such as high altitude or obstructive sleep apne,17,18 it is important to note that oxygen levels in periodontal pockets remain inherently low even in common forms of periodontitis.19 This may explain the minimal microbiome alterations observed in this study.
To further dissect microbiome contributions, we eliminated dental plaque using antibiotic-treated ligatures (Fig. S1d). Despite plaque elimination, periodontitis symptoms remained aggravated under hypoxia compared to antibiotic-treated mice in NO conditions (Fig. S1e–g). These findings suggest that the oral microbiome is not the primary mediator of hypoxia-aggravated mucosal immunopathology. Instead, hypoxia may exacerbate periodontitis through alternative mechanisms.
Role of NETosis in exacerbating oral mucosal lesions under hypoxiaProgression of oral mucosal immunopathology is not solely related to the oral microbiome; it is also influenced by a dysfunctional immune response. Having excluded microbiome changes, we explored the the role of the immune system in hypoxia-aggravated mucosal immunopathology. Mice were subjected to LIP and housed in either low-oxygen (LO-LIP) or normal-oxygen (NO-LIP) environments. Gingival tissues derived from these mice that underwent RNA sequencing revealed significant gene expression differences between the LO-LIP and NO-LIP groups (Fig. 2a). Gene ontology analysis showed a marked upregulation of pathways related to neutrophil activation and neutrophil-mediated inflammatory responses in LO-LIP mice (Fig. 2b).
Fig. 2
The effect of NETs on aggravates periodontitis under hypoxic condition. a Heatmap of the differential genes. b Enriched gene ontology terms comparing LO-LIP and NO-LIP groups. c Flow cytometry analysis of gingiva tissues from LO-LIP and NO-LIP mouse. d, e Absolutely count of neutrophils (d) and monocytes (e) in mouse gingiva (n = 6). f Representative CLSM images of periodontal tissues from LO-LIP and NO-LIP mice which were stained with Ly6G and DAPI. Scale bars, 100 μm. g Semi-quantitative analysis of Ly6G signaling in e (n = 6). h Concentration of NE in the lysate of mouse periodontal tissue (n = 6). i GSEA plot of the upregulated NETs formations KEGG pathways in LO-LIP groups. j Representative CLSM images of periodontal tissues from LO-LIP and NO-LIP mice, which was stained with cit H3 and DAPI. Scale bars, 100 μm. k Semi-quantitative analysis of cit H3 signaling in (i) (n = 6). Concentration of MPO-DNA complex in the lysate of mouse periodontal tissue (n = 6) (l) and the GCF of periodontitis patients (n = 20) (m). n Schematically illustrating the design of in vivo experiments. o Representative images of H&E staining (left), TRAP staining (middle) and micro-CT (left) of the periodontal tissue of Padi4-/- mice and wild type mice. The red dash represents the boundary between bone and connective tissue. The yellow dash represents the boundary between connective tissue and epithelium. Scale bars of H&E staining and TRAP staining, 200 μm. Scale bars of micro-CT, 1 mm. p Semi quantitative analysis of TRAP staining in (o) (n = 6). q Bone loss measurement of Padi4−/− mice and wild type mice (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001; **P < 0.01; *P < 0.05; ns, no significance. Statistical significance was determined by two-tailed unpaired Student’s t test. NETs neutrophil extracellular traps, MPO-DNA myeloperoxidase-deoxyribonucleic acid, CLSM confocal laser scanning microscopy, GCF gingival crevicular fluid, NE neutrophil elastase, cit H3 citrullinated histone H3, H&E hematoxylin and eosin, LIP ligature-induced periodontitis, TRAP Tartrate resistant acid phosphatase, micro-CT micro-computed tomography, LO low oxygen, NO normal oxygen)
Consistent with transcriptomic data, flow cytometry demonstrated increased neutrophil infiltration (live CD45+CD11b+Ly6G+) in both early and late stages of gingival inflammation in LO-LIP mice (Fig. 2c, d). In contrast, other inflammatory cells including monocytes (live CD45+CD11b+Ly6G-), B cells (live CD45+B220+), and T cells (live CD45+TCRβ+) only demonstrated increased infiltration at later stages (Fig. 2e, Fig. S2). Immunostaining for Ly6G and elevated neutrophil elastase further validated these findings (Fig. 2f–h). Collectively, these findings suggest that neutrophils play a central role in exacerbating periodontitis under hypoxic conditions. However, the precise mechanism by which neutrophils contribute to disease progression requires further investigation.
Further analysis of RNA-seq data was conducted to examine the role of neutrophils in hypoxia-aggravated periodontitis. Gene Set Enrichment Analysis revealed an upregulation of NET formation in LO-LIP mice (Fig. 2i). LO-LIP mice showed elevated NET markers-citrullinated histone H3 (citH3) and myeloperoxidase (MPO)-DNA complexes in gingival tissues (Fig. 2j–l). This finding was also confirmed in human GCF samples (Fig. 2m). These results suggest that NETosis plays an indispensible role in the exacerbation of periodontitis under hypoxic conditions. To functionally validate NETosis’ role, we used Padi4−/− mice, peptidyl arginine deiminase type 4 (Padi4)-deficient mice (Padi4−/−) that lack the ability to form NETs were used to validate this hypothesis. Following a 14-day exposure to LIP in LO environment (Fig. 2n), Padi4−/− mice exhibited significantly reduced periodontitis severity (Fig. 2o, p). Degradation of existing NETs using DNase I also alleviated periodontitis under hypoxia (Fig. S3). These findings indicate that excessive NET formation under hypoxic conditions contributes significantly to oral mucosal inflammation. However, the precise mechanisms driving increased NETosis in hypoxia remain to be elucidated.
Effect of hypoxia on platelet-induced NETosisBacteria are potent stimuli for NET formation in common forms of periodontitis.10 Periodontal pathobionts such as Porphyromonas gingivalis (Pg), have been shown to directly induce NET formation.20,21 However, oral microbiome specimens from LO-LIP and NO-LIP mice showed no significant differences in their ability to elicit NET formation (Fig. S4). This prompted further investigation into whether hypoxia directly induces NET release and contributes to periodontitis progression.
To examine this notion, neutrophils from mice adapted to an LO environment for 7 days (LO-Neu) and normoxic controls (NO-Neu) were collected and cultured in a medium containing inactivated Pg. The aforementioned experiment was conducted to simulate the periodontitis microenvironment in vitro. Both LO-Neu and NO-Neu were incubated under hypoxic and normoxic conditions, respectively. Evaluation of NET formation indicated no significant differences between the two groups (Fig. S5a, b), suggesting that hypoxia alone does not directly enhance NET formation.
The potential role of hypoxia-exposed neutrophils (LO-Neu) directly exacerbates periodontitis was further examined in an in vivo model. This model comprised using mice that express the diphtheria toxin receptor (DTR) under the control of the Ly6G promotor that is expressed specifically in neutrophils (Ly6G-DTR mice) to achieve neutrophil depletion.22 Host NO-Neu were depleted before subjecting the mice to LIP under normoxic conditions. Daily DT injections were used to deplete endogenous neutrophils, and LO-Neu or NO-Neu from donor mice was transfused to evaluate their effects on periodontitis (Fig. S5c). The results showed that LO-Neu failed to aggravate periodontitis compared to NO-Neu (Fig. S5d–f). This observation further indicates that hypoxia does not directly exacerbates neutrophil-mediated periodontitis.
Platelet activation plays a critical role in the pathogenesis of hypoxia-related diseases and contributes to NET formation.12,13,15 Hence, it is logical to examine whether platelets respond to hypoxia by upregulating NET formation and exacerbating oral mucosal inflammation. Microarray analysis of blood samples from healthy adults transported to high altitudes revealed upregulation of pathways related to platelet aggregation.23 Further RNA sequencing analysis in the present study confirmed increased expression of genes associated with platelet activation (Fig. 3a). Consistent results observed in GCF samples collected from periodontitis patients reinforced the presence of platelet activation-related gene signatures (Fig. 3b). These findings suggest that platelet activation may contribute to periodontitis exacerbation under hypoxic conditions. Increased platelet and neutrophil infiltration in the gingival tissue of LO-PLT mice further indicated a potential interaction between these cell types (Fig. 3c). Flow cytometry analysis demonstrated a high percentage of neutrophil-platelet aggregation in the blood of LO-LIP mice (Fig. 3d, e) and higher percentage of activated platelets from LO-LIP mice (Fig. 3f, g). These findings suggest that hypoxia may influence platelet activation and promote neutrophil-platelet aggregation.
Fig. 3
Platelets mediated NETosis upregulation under hypoxic condition. a Heatmap of representative differentially expressed genes related to platelet activation. b Concentration of P-selectin in the GCF of periodontitis patients (n = 20). c CLSM images of periodontal tissues from LO-LIP and NO-LIP mice, which were stained with Ly6G (red) and CD41 (green). Scale bars, 100 μm. d Flow cytometry analysis of neutrophil-platelet aggregation. e Changes of neutrophil-platelets aggregation in percentage (n = 6). f Flow cytometry analysis of P-selectin positive platelets. g Changes of P-selectin positive platelets in percentage (n = 6). h, j Representative CLSM (h) and SEM (j) images of neutrophil incubated with LO-PLT and NO-PLT for 2 h, 4 h, 6 h, and 8 h. Scale bars of CLSM, 50 μm. i Semi-quantitative of cit H3 signaling in (h) (n = 6). k Concentration of MPO-DNA complex in the supernatant of neutrophils incubated with LO-PLT or NO-PLT (n = 6). l Schematic illustrating the design of in vivo experiments. m Representative TRAP staining and micro-CT images of the periodontal tissue of PF4-DTR mice transfused with LO-PLT and NO-PLT. Scale bars of TRAP staining, 200 μm. Scale bars of micro-CT 1 mm. n Semi-quantitative analysis of TRAP staining in (m) (n = 6). o Bone loss measurement of differently treated PF4-DTR mice (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001; **P < 0.01. Statistical significance was determined by two-tailed unpaired Student’s t test. MPO-DNA myeloperoxidase-deoxyribonucleic acid, CLSM confocal laser scanning microscopy, GCF gingival crevicular fluid, NE neutrophil elastase, PLT platelets, cit H3 citrullinated histone H3, TRAP Tartrate resistant acid phosphatase, LIP ligature-induced periodontitis, micro-CT micro-computed tomography, LO low oxygen, NO normal oxygen, Neu neutrophils
To examine whether increased neutrophil-platelet aggregation enhances NET formation, platelets from LO-LIP and NO-LIP mice were incubated with neutrophils in vitro. Over time, LO-PLT significantly promoted NET formation (Fig. 3h–k), suggesting that hypoxia may drive NETosis through platelet activation. To directly test platelet involvement, platelet Factor 4 (PF4)-DTR mice were used to further confirm the role of LO-PLT in exacerbating periodontitis under hypoxic conditions.24 Platelets were depleted using DT, after which the mice underwent LIP in a hypoxic environment and received transfusions of either LO-PLT or NO-PLT (Fig. 3l). The PF4-DTR mice transfused with NO-PLT showed alleviated periodontitis symptoms (Fig. 3m–o). Taken together, these observations indicate that hypoxia upregulated platelet activation, enhances neutrophil-platelet aggregation, and drives NETosis to exacerbatr periodontitis. Further research is needed to elucidate the precise mechanisms by which hypoxia induces platelet activation and NET formation.
Contribution of hypoxia to platelet cGAS-STING signaling and NET formationIn the procedure of platelets activation mediated NETs formation, the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-stimulator of interferon genes (STING) pathway plays a profound role.25 To be specifically, the product of cGAS activation-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) induces palmitoylation of STING, enabling its interaction with syntaxin binding protein 2 (STXBP2) to mediate platelet activation and NET formation.4 Although previous study have linked cGAS-STING pathway activation to hypoxia-induced disease, whether hypoxia upregulate cGAS-STING pathway and by which mechanism remain obscure.9 Therefore, we hypothesize that hypoxia exacerbates cGAS-STING activation in platelets, thereby enhancing downstream effects such as NET formation.
Evaluation of cGAS-STING pathway activation in LO-PLT and NO-PLT revealed a significant increase in cytosolic levels of cGAMP, the product of cGAS, in platelets exposed to hypoxia (Fig. 4b). Consistent with this observation, LO-PLT exhibited elevated STING palmitoylation and enhanced binding to syntaxin binding protein 2 (Fig. 4c–f). These findings suggest that hypoxia enhances activation of the platelet cGAS-STING pathway.
Fig. 4
Hypoxia amplifies platelet cGAS-STING signaling to enhance NETs formation. a Schematic illustration of the overall mechanism in this figure. b Concentration of cGAMP in the cytoplasm from 108 platelets (n = 6). c The level of STING palmitoylation in LO-PLT and NO-PLT after incubated with Pg medium. d Quantitative analysis of the gray values of STING palmitoylation in (c) (n = 6). e IP-STING assay of LO-PLT and NO-PLT after incubated with Pg medium. Isotype-matched IgG served as the negative control. f Quantitative analysis of STBQX2 bands in (e) (n = 6). g Flow cytometry analysis of P-selectin positive platelets. h Changes of P-selectin positive platelets in percentage (n = 6). i Representative CLSM images of neutrophils incubated with C-ST5 treated LO-PLT or untreated LO-PLT. Scale bars, 50 μm. j Semi-quantitative of cit H3 signaling in (i) (n = 6). k Concentration of MPO-DNA complex in the supernatant of neutrophils incubated with C-176 treated HO-PLT or untreated HO-PLT (n = 6). l Expression of cit H3 in neutrophils incubated with C-ST5 treated LO-PLT or untreated LO-PLT. m Semi-quantitative analysis of cit H3 in (l) (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001; **P < 0.01; ns, no significance. Statistical significance was determined by two-tailed unpaired Student’s t test. MPO-DNA myeloperoxidase-deoxyribonucleic acid, CLSM confocal laser scanning microscopy, cit H3 citrullinated histone H3, PLT platelets, LO low oxygen, NO normal oxygen, Neu neutrophils, cGAMP cyclic guanosine monophosphate-adenosine monophosphate, HAM hydroxylamine, STING stimulator of interferon genes, STXBP2 syntaxin binding protein 2
To further elucidate the functional role of the cGAS-STING pathway in platelet activation under hypoxic conditions, the C-ST5 peptide, a platelet-specific inhibitor of this pathway, was employed. Inhibition of STING binding to syntaxin binding protein 2 with C-ST5 significantly reduced LO-PLT activation (Fig. 4g, h) and concurrently weakened NET formation (Fig. 4i–m). These results highlight the role of the cGAS-STING pathway in platelet activation and NET formation under hypoxia. This prompted further investigation into the mechanisms driving cGAS-STING upregulation in platelets under hypoxic conditions.
Effect of hypoxia on mtDNA metabolism in plateletscGAS functions as a cytosolic DNA sensor and is activated by elevated levels of double-stranded DNA (dsDNA) in the cytoplasm.26 Consistent with this mechanism, an increased concentration of cytosolic DNA was consistently observed in LO-PLT (Fig. 5b), which may contribute to abnormal cGAS activation. Platelets contain both endogenous and exogenous dsDNA, with endogenous dsDNA originating from mitochondria (mtDNA) and exogenous dsDNA derived from ingested fragments of nucleated cells or bacteria.27 Therefore, identifying the source of the increased cytosolic DNA was necessary.
Fig. 5
TFAM-dependent cytosolic mtDNA degradation is inhibited in platelets under hypoxic condition. a Schematic illustration of the overall mechanism in this figure. b Concentration of dsDNA in the cytoplasm from 108 platelets (n = 6). c Images from agarose gel electrophoresis of the DNA extracted from cytoplasm of LO-PLT and NO-PLT incubated with Pg medium. Mitochondrial DNA isolated from platelets, nuclear DNA isolated from neutrophils and Pg DNA isolated from Pg were used as controls. d CLSM images of LO-PLT and NO-PLT incubated with Pg medium, stained with DNA (green) and TOM20 (red). Scale bars, 5 μm. e, f Semi-quantitative of the DNA signaling not merged with TOM20 (e) and the DNA signaling merged with TOM20 (f) in (d) (n = 6). g, h Quantification of mtDNA copy number by PCR using the probe of Atp6, Co1, Nd1 and D-loop, from isolated cytosolic fractions (g) or mitochondrial fractions (h) of LO-PLT and NO-PLT incubated with Pg medium (n = 6). i CLSM images of LO-PLT and NO-PLT incubated with Pg medium, stained with DNA (green), TFAM (red) and TOM20 (blue). Scale bars, 5 μm. j Semi-quantitative of the DNA signaling that merged with TFAM in (l) (n = 6). k Chromatin immunoprecipitation assay was conducted to examine the occupancy of TFAM within mtDNA promoter regions, including HSP1, HSP2, and LSP, in LO-PLT and NO-PLT incubated with Pg medium (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001; **P < 0.01; ns, no significance. Statistical significance was determined by two-tailed unpaired Student’s t test (b, e, f, g, h, j) or two-way ANOVA test (k). CLSM confocal laser scanning microscopy, LO low oxygen, NO normal oxygen, PLT platelets, TFAM mitochondrial transcriptional factor A, LC3B microtubule-associated protein 1 light chain 3, mtDNA, Pg Porphyromonas gingivalis, TOM20 translocase of outer mitochondrial membrane 20, mtDNA mitochondrial DNA
To elucidate the source of the increased cytosolic DNA, DNA was extracted from platelets, amplified, and analyzed using polymerase chain reaction (PCR). Agarose gel electrophoresis revealed that cytosolic DNA in LO-PLT and NO-PLT primarily originated from mitochondria rather than external sources (Fig. 5c). These findings explain the upregulation of the cGAS-STING pathway and the accumulation of excess cytosolic DNA. However, the mechanism by which hypoxia mediated the increase of cytosolic mtDNA in platelets requires further investigation.
Given that platelets are anucleate, the increased concentration of mtDNA in their cytoplasm may arise from either enhanced mtDNA leakage from mitochondria or impaired mtDNA degradation. Immunofluorescence analysis showed no significant changes in mitochondrial DNA content, as indicated by the colocalization of red and green fluorescent signals (Fig. 5d, e). However, an increase in cytosolic DNA was observed in LO-PLT, represented by non-co-localized green signals (Fig. 5d, f). These observations were further confirmed by quantitative PCR (Fig. 5g, h).
Moreover, mitochondrial reactive oxygen species (ROS), which are implicated in mtDNA leakage in activated platelets, showed no significant differences between LO-PLT and NO-PLT.28 Furthermore, the integrity of mtDNA in LO-PLT was also comparable to that of the control group (Fig. S6). These results indicate that the accumulation of cytosolic mtDNA is not attributable to mitochondrial leakage but rather to impaired mtDNA degradation under hypoxic conditions.
Cytosolic mtDNA degradation is regulated by two distinct pathways. The first involves non-specific degradation mediated by three prime repair exonuclease 1 (TREX1), while the second relies on an autophagy pathway driven by mitochondrial transcriptional factor A (TFAM).29,30 However, the expression levels of both TREX1 and TFAM remained unchanged between LO-PLT and NO-PLT (Figure. S7).
Since TFAM-dependent mtDNA degradation requires its binding to mtDNA,30 immunofluorescence staining was performed to examine TFAM and DNA co-localization in platelets. The results showed a significant reduction in TFAM-mtDNA co-localization in LO-PLT (Fig. 5i, j), suggesting impaired mtDNA degradation in the cytoplasm. This observation was further supported by chromatin immunoprecipitation, which confirmed a weaker interaction between TFAM and mtDNA in LO-PLT (Fig. 5k).
These findings reveal a mechanism underlying impaired cytosolic mtDNA degradation in platelets under hypoxic conditions: TFAM dissociation from mtDNA disrupts the TFAM-mediated degradation pathway. Consequently, the accumulation of cytosolic mtDNA upregulates the cGAS-STING signaling pathway to enhance platelet activation and NET formation (Fig. 5a). Further investigation is needed to determine how hypoxia disrupts TFAM-mtDNA binding.
Two factors inhibit the interaction between mtDNA and TFAM. The first factor is mtDNA modification, specifically N6-methyladenine (6 mA). This is the only known modification that interferes with TFAM binding to mtDNA to interfere with its degradation.31 The second factor involves TFAM modifications such as phosphorylation and acetylation.32 However, studies on TFAM modifications have yielded conflicting results. Whereas one study reported that phosphorylated or acetylated TFAM has reduced affinity for mtDNA in vitro,32 a more recent study found that these modifications do not affect TFAM-mtDNA binding.33
To address these discrepancies, the impact of hypoxia on mtDNA 6 mA levels in platelets was investigated. The significant increase in mtDNA 6 mA in LO-PLT (Fig. 6b–d) suggests that elevated 6 mA methylation may inhibit mtDNA binding to TFAM under hypoxia. Methyltransferase-like protein 4 (METTL4), a eukaryotic homolog of the bacterial 6 mA methyltransferase DNA adenine methyltransferase 1, is known to catalyze 6 mA methylation in mammalian DNA.31 A previous study reported that METTL4 expression is significantly upregulated through p53 activation when cardiomyocytes are exposed to hypoxia.34 A similar mechanism may occur in platelets. Indeed, METTL4 was specifically localized to platelet mitochondria (Fig. 6e), and its expression was significantly upregulated in LO-PLT (Fig. 6e–h). These findings indicate that hypoxia upregulated METTL4 in platelets, facilitates mtDNA 6 mA methylation, disrupts TFAM-dependent mtDNA degradation, and results in cytoplasmic mtDNA accumulation (Fig. 6a).
Fig. 6
Hypoxia promotes mtDNA 6 mA methylation via METTL4 in platelets. a Schematic illustration of the overall mechanism in this figure. b CLSM images of LO-PLT and NO-PLT stained with 6 mA (red) and sybr green (green). Scale bars, 5 μm. c Semi-quantitative of the 6 mA signaling in (b) (n = 6). d mtDNA 6 mA levels of LO-PLT and NO-PLT were assessed by dot blot analysis. e CLSM images of LO-PLT and NO-PLT stained with METTL4 (green) and TOM20 (red). Scale bars, 5 μm. f Semi-quantitative of the 6 mA signaling in (e) (n = 6). g The expression of METTL4 in LO-PLT and NO-PLT. h Semi-quantitative of METTL4 in (g) (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001. Statistical significance was determined by two-tailed unpaired Student’s t test. CLSM confocal laser scanning microscopy, LO low oxygen, NO normal oxygen, PLT platelets, mtDNA mitochondrial DNA, 6 mA N6-methyladenine, LC3B microtubule-associated protein 1 light chain 3, METTL4 methyltransferase-like protein 4
Impact of platelet METTL4 depletion on periodontitis in hypoxiaTo elucidate the role of METTL4 in mtDNA degradation within platelets, platelet METTL4-deficiency mice (Mettl4f/fPf4-Cre+, Mettl4-/-) and their littermate control mice (Mettl4f/fPf4-Cre-, Mettl4f/f) were used to clarify the role of METTL4 in mtDNA degradation within platelets (Fig. S8a, b). In Mettl4−/− mice, the protein level of METTL4 in platelets were significantly decreased in platelets compared to that in leukocytes and erythrocytes (Fig. S8c). LO-PLT specimens were obtained from both Mettl4−/− and Mettl4f/f mice for evaluating the cytosolic DNA concentration. There was a significant reduction in cytosolic DNA concentration in LO-PLT from Mettl4−/− mice (Fig. 7a). Flow cytometry analysis demonstrated a high percentage of neutrophil-platelet aggregation in the blood of LO-LIP mice (Fig. 7b, c). Additionally, the formation of NETs was also reduced in neutrophils co-cultured with LO-PLT from Mettl4−/− mice (Fig. 7d, e). These findings prompted further investigation in whether METTL4 is a potential target for mitigating periodontitis exacerbation under hypoxic conditions.
Fig. 7
Platelet METTL4 deficiency alleviates periodontitis under hypoxic condition. a Concentration of cytosolic dsDNA in LO-PLT derived from Mettl4−/− and Mettl4f/f (n = 6). b Flow cytometry analysis of P-selectin positive platelets. c Changes of P-selectin positive platelets in percentage (n = 6). d CLSM images of neutrophils incubated with LO-PLT derived from Mettl4−/− and Mettl4f/f. Scale bars, 50μm. e Semi-quantitative of the cit H3 signaling in (d) (n = 6). f Schematic design of the in vivo experiment. g Representative CLSM images of periodontal tissues of Mettl4−/− and Mettl4f/f mice that subjected to LO-LIP, stained with cit H3 and DAPI. Scale bars, 100 μm. h Semi-quantitative of the cit H3 signaling in (g) (n = 6). i Relative gingival mRNA expression of inflammatory cytokines of Mettl4−/− and Mettl4f/f mice that subjected to LO-LIP (n = 6). j, l Representative images of H&E staining (left), TRAP staining (right) and micro-CT (l) of the periodontal tissue of Mettl4−/− and Mettl4f/f mice that subjected to LO-LIP. The red dash represents the boundary between bone and connective tissue. The yellow dash represents the boundary between connective tissue and epithelium. Scale bars of H&E and TRAP staining, 200 μm. Scale bars of micro-CT, 1 mm. k Semi-quantitative analysis of TRAP staining in (j) (n = 6). m Bone loss measurement of differentially treated mouse (n = 6). The data are presented as the mean ± SD. ****P < 0.000 1; ***P < 0.001; **P < 0.01. Statistical significance was determined by two-tailed unpaired Student’s t test. CLSM confocal laser scanning microscopy, LO low oxygen, NO normal oxygenm PLT platelets, mtDNA mitochondrial DNA, METTL4 methyltransferase-like protein 4, dsDNA double-stranded DNA, cit H3 citrullinated histones H3, TRAP Tartrate resistant acid phosphatase, LIP ligature-induced periodontitis, H&E hematoxylin and eosin, micro-CT micro-computed tomography
Mettl4−/− mice and Mettl4f/f mice were subsequently subjected to LIP and raised in hypoxic conditions (Fig. 7f). In the Mettl4−/− mice, NET formation was significantly inhibited in the gingival tissue (Fig. 7g, h). This was accompanied by downregulated expression of inflammatory genes in the gingiva (Fig. 7i). In line with these results, the Mettl4−/− mice exhibited alleviated symptom of periodontitis (Fig. 7j–m). These findings demonstrate that platelet METTL4 deficiency mitigates hypoxia-driven periodontitis exacerbation.
Comments (0)