Resource availability
Further information and requests for resources and reagents, please contact Junfa Yuan and will be fulfilled (jfyuan@mail.hzau.edu.cn).
Materials availability
This study did not generate new unique materials.
Data and code availability
Data: All data reported in this paper will be shared by the lead contact upon request.
Code: This paper does not report original code.
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant detailsGene identification and bioinformatics
A query set of available Oncorhynchus mykiss (IFNs) and Danio rerio (IFNs) sequences was used to search the whole genome [ASM336829v1] and transcriptome [PRJNA977193] of Cauratus gibelio were searched using the BLASTN and TBLASTX procedures to inquire about the homologous genes (IFNs). Sequences were confirmed by quantitative PCR (qPCR), and the primers were attached in the Tables S1. ORF Finder (http://www.bioinformatics.org/sms/orffind.html) was used to predict genes, and ExPASy's translate tool (http://web.expasy.org/translate/) was used to translate proteins. The software SignalP (http://www.cbs.dtu.dk/services/SignalP/) was used to examine signal peptide predictions, whereas conserved domains (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) was used to predict the DBD domains of IFNs. Multiple alignments were calculated via the CLUSTAL W program, and phylogenetic trees were built with the MEGA (version 7) software tool utilizing the minimum-evolution and neighbor-joining methods. The full-length cDNA of CaIFNυ was obtained by 5’and 3’RACE PCR from intestinal tissue (5’- ATGTTCTGGATTAGGATCA-3’, 3’-ACGCCGTCCTCTGACAGAG-5’), and its promoter sequence was obtained through the NCBI database. The transcription factor binding sites of CaIFNυ in its promoter were predicted by animal TFBD 4.0 (bioinfo.life.hust.edu.cn) and mapped with IBS (Illustration for Bio Sequence).
Construction of the structural model
The predicted structures of CaIFNυ, CaCRFB4, and CaCRFB12 were constructed using the Swiss model (https://swissmodel.expasy.org/). CaIFNυ, CaCRFB4, and CaCRFB12 models were generated by PyMOL software. Zdock software was used to analyze the possibility of docking analysis between CaIFNυ, CaCRFB4, and CaCRFB12. The prediction was further analyzed using PDBePISA: Protein Interaction Analysis Software (https://www.ebi.ac.uk/pdbe/pisa/).
Fish, cell lines and infectious agents
Fish
The fish were purchased from a nearby commercial farm in Wuhan, Hubei, China, and were found to be in good health, weighing an average of 100 ± 4.31 g. They were kept in a holding tank (0.8 m2 × 1.2 m) with an aerated carbon filter, dechlorinated tap water, and a natural photoperiod for 14 days prior to the experiment. The temperature was kept between 23 ~ 25 °C. The Scientific Ethic Committee of Huazhong Agricultural University (HZAUFI-2022–016) approved all animal studies, and all fish utilized in this study were kept and cared for at HZAU in compliance with institutional animal care.
Cell lines
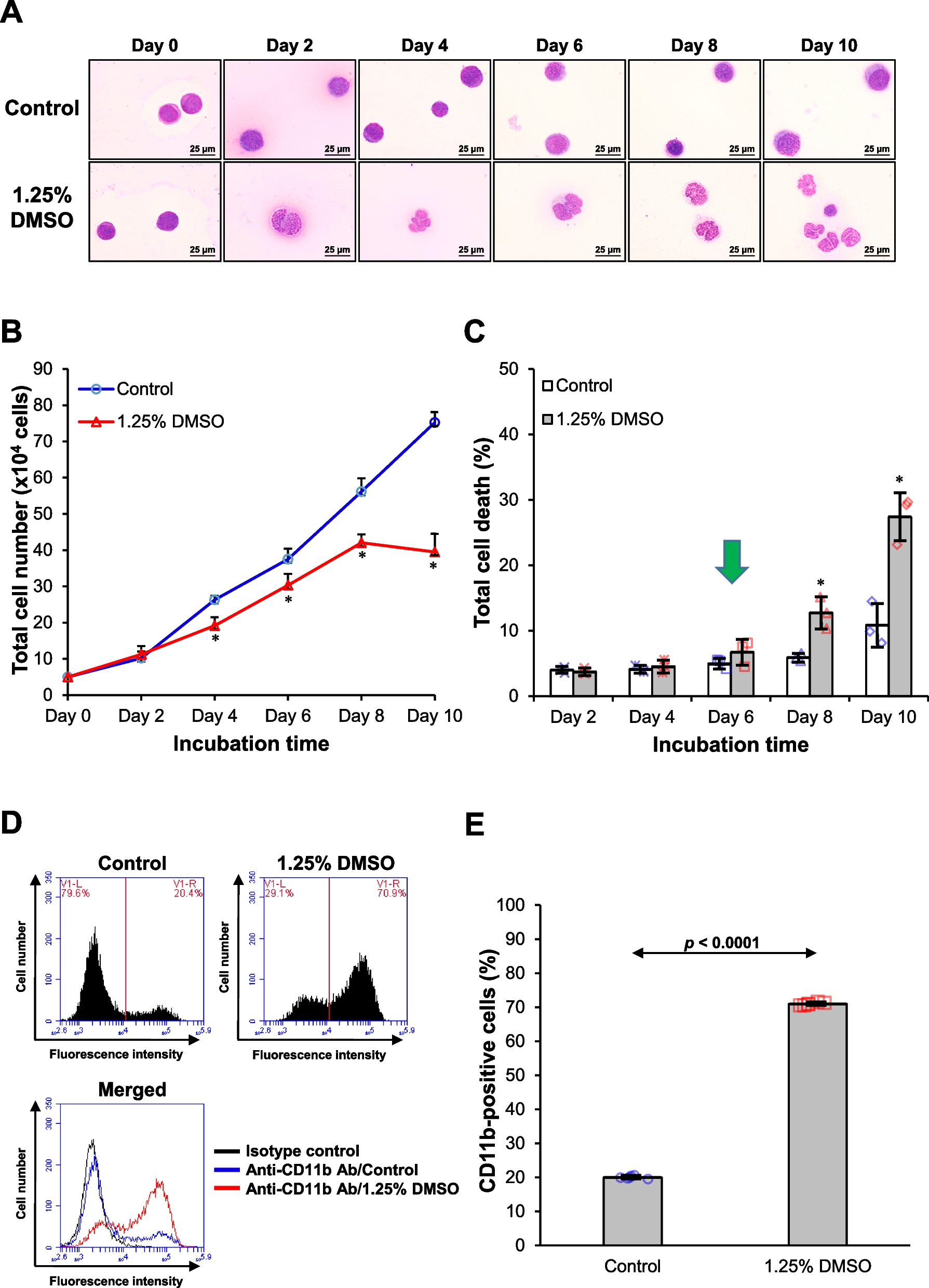
Fathead minnow cell line, FHM cells were grown in M199 medium (Gbico, American) supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher) at a temperature of 25 °C and a CO2 atmosphere of 5%. The cells were preserved in the laboratory, and the results of bioinformatics analysis showed that FHM cells could be used in this experiment (Figure S3-S6). Professor Huajun Zhang of the Wuhan Institute of Virology, Chinese Academy of Sciences kindly donated human embryonic kidney 293 T (HEK293T, GDC0187) cells, which were cultured in DMEM (Gbico, American) supplemented with 10% FBS (Gbico, American) at 37 °C in an incubator with 5% CO2 (Gbico, American).
Infectious agents
The Cyprinid herpesvirus II (CyHV-2) isolate of SY-C1 was stored in the laboratory at −80°C [49]. The Spring viraemiaof carp virus (SVCV) was also stored in the laboratory at −80°C.Viral copies were determined by qPCR according to the previously report [50]. Aeromonas hydrophila strain (Ah541), previously isolated from Carassius gibelio, was used in this experiment and counted colony-forming units (CFU) on LB culture (LB) plates for serial dilutions [51]. The median lethal dose (LD50) of CyHV-2 or A. hydrophila was determined by artificial infection with C. gibelio.
CyHV-2 and A. hydrophila challenge
Fish were infected intraperitoneally with 100 μl virus stock containing 1 × 105.5 copies/ml CyHV-2 (0.1 × LD50). In short, 10 ml of TN buffer (100 mmol/l NaCl, 50 mmol/l Tris–HCl, pH 7.4) was used to combine and homogenize 1 g of spleen tissues from C. gibelio infected with CyHV-2. The samples were then centrifuged at 4 °C for 10 min. The supernatant was filtered through an acetate cellulose filter measuring 0.45 μm and injected into healthy C. gibelio. Fish in the control group were injected with the same volume of TN buffer. For bacterial infection, healthy fish were intraperitoneally injected with 200 μl of A. hydrophila in phosphate-buffered saline (PBS, 108 CFU/ml), and control fish was intraperitoneally injected with 200 μl of PBS. At 0, 3, 6, 12, 24, 72, and 168 h after infection (hpi), tissues from the infected and control groups were taken, including the spleen, midgut, and head kidney. After being frozen in liquid nitrogen, the samples were moved to a temperature of 80°C.
Total DNA/RNA extraction, cDNA synthesis and quantitative PCR
Following the manufacturer's instructions, DNA and total RNA were extracted using TRIzol (Cwbio, China) and a tissue DNA purification kit (CW2298, CWBio, China), respectively. RNA quality and concentrations were assessed using NanoDrop 2000 (Thermo Scientific, America). A 1 μg aliquot of total RNA was used for reverse transcription with the HiScript® reverse transcriptase (Vazyme Biotech, China) to convert total RNA into cDNA. 20 µl reactions were used for the quantitative PCR, with 0.5 µl of each indicated primer and 10 µl of AceQ® qPCR SYBR Green Master Mix. The cycle parameters for the amplification reactions were as follows: 94 °C for 5 min; 35 cycles of 96 °C for 30 s, 60 s at 56 °C, 90 s at 72 °C, and 72 °C for 10 min. Table S1 provides specifics on the qPCR primers used in this investigation. Specifically, the data were analyzed using the standard 2 − ΔΔCT(Livak) method, with β-actin serving as an endogenous control to standardize gene expression. Graphpad Prism 7.0 software was utilized to conduct statistical analysis on the data, which were presented as the mean ± standard deviation.
Plasmid construction
The CaIFNa1 (XM_026209886.1), CaIFNυ (XM_026200839.1), CaIRF3 (XM_026277295.1), and CaIRF7 (XM_026239392.1) were amplified using gene-specific primers (Table S1) and inserted into the indicated vectors, respectively. Plasmids pcDNA4.0-CaIFNa1, pcDNA4.0-CaIFNυ, pEGFP-N1-CaIFNυ, pCMV-flag-CaIRF3, pcDNA4.0-CaIRF7, and pEGFP-N1-CaNF-κB(p65) were constructed. The following gene fragments were subcloned to create plasmids for coimmunoprecipitation (CO-IP): XM_026272783.1, which encodes the extracellular region of CaCRFB4 (pcDNA4.0-CaCRFB4), and XM_026212441.1, which encodes the extracellular region of CaCRFB12 (pcDNA3.1-Myc-CaCRFB12). The plasmid of pcDNA3.1-STAT1a was a kind gift from professor Zou Jun (Shanghai Ocean University, Shanghai, China) and details of this plasmid could found in the report [52], and the plasmid pEGFP-N1-STAT1a is the result of changing the Tag the pcDNA3.1-STAT1a. The plasmid of pEGFP-N1-IRF1 was a kind gift from professor Yibing Zhang (Institute of Hydrobiology, Chinese Academy of Sciences). The promoter regions of CaIFNa1 (ASM336829v1, 12049010 ~ 12051010) and CaIFNυ (ASM336829v1, 6672614 ~ 6675614) were amplified and inserted into the pGL3-basic vector. All the plasmids were extracted and purified by using a plasmid Maxi Kit (Promega, America) following a standard protocol.
Recombinant protein expression in HEK293T cells
Following the manufacturer's instructions, HEK293T cells were transfected with plasmid using LipofectamineTM 3000 Transfection Reagent (Invitrogen, America) after being grown in a T75 cell culture flask at a concentration of 3 × 106 cells/ml.
Briefly, 25 µg of the pcDNA4.0-CaIFNa1 or pcDNA4.0-CaIFNυ plasmid was combined with 1.5 ml of Opti-MEMTM reduced serum medium (Gibco, America). Subsequently, 75 µl of LipofectamineTM 3000 Transfection Reagent was pipetted into a solution. After an extra 25 min at room temperature, the solutions were introduced to the flasks holding the 293 T cells. After 48 h of cell culture in 5% CO2 at 37 °C, we removed the culture medium and used Ni-IDA SefinoseTM Resin (Sangon Biotech, China) to purify the recombinant proteins. Western blotting and SDS-PAGE were used to confirm the purified proteins. The BCA Protein Colorimetric Assay Kit was used to measure the protein concentration.
Luciferase reporter assay
The luciferase reporter assay was performed as previously reported [53]. Briefly, FHM cells were plated in 12-well plates (1 × 105 cells/well) and then co-transfected with 1 μg of CaIFNa1 or CaIFNυ reporter plasmids with different dosages (0, 50, 100, 150, 200 ng) of IRF1-, IRF3-, IRF7-, and p65-expressing plasmids, respectively. 100 ng pRL-TK was transfected as an internal control. After 24 h of transfection, the cells were harvested for detection of luciferase activity. According to the previous [23], the cooperativity analysis between any two of IRF1/3/7 on CaIFNa1 and CaIFNυ promoter activation was computed. Plated in 12-well plates (1 × 105 cells/well), FHM cells were co-transfected with the CaIFNa1 or CaIFNυ promoters (1 μg) in the presence of two IRF plasmids at the given concentrations. As an internal control, 100 ng of pRL-TK was added. After that, the cells were harvested 24 h later, and the Dual Luciferase Assay System (Promega; E1960) was used to detect them.
Viral infection and viral plaque assay
FHM cells were cultivated on a 6-well plate at 1 × 106 cells/well, and then treated with CaIFNa1 or CaIFNυ recombinant proteins at 60 ng/ml for 12 h. Subsequently, each well was infected with 200 μl of virus-containing supernatants (MOI = 0.1) and allowed to adsorp for 60 min. Each 6-well plate replaced with 2 ml of sustaining media containing 5% FBS. The cells were then kept at 28°C. After 24 h of treatment, the supernatant was collected to determine the viral titers. For viral plaque assay,
after seeding FHM cells in a 12-well plate (1 × 105/cm2) for 12 h, 100 μl of the virus-containing supernatants solution was treated at tenfold serial dilutions before being inoculated into three duplicates of the cells. Following an hour of absorption, the cells were cultivated for 72 h in M199 supplemented with 2% sodium carboxymethyl cellulose and 5% foetal bovine serum. The cells were fixed for 6 h in 10% formaldehyde, and then stained for an hour with 0.5% crystal violet. Following a rinse with tap water, the formula PFU/ml = 10a × b × (1,000/c) was used to calculate the number of visible plaques. Here, a represents the number of plaques, b is the amount of virus-containing supernatant diluted times, and c represents the volume of virus-containing supernatant.
Western blots and fluorescence confocal
Protein expression was determined using Western blots. To prepare for western blotting, the cell treated with SVCV or the transfected cell for pEGFP-N1-STAT1a or pcDNA3.1-STAT1a were gathered and lysed using RIPA buffer (Liuyi Biotechnology, China). After that, it was blocked for 2 h at 37 °C using TBS buffer containing 3% skim milk, and it was then treated for 2 h at room temperature with the primary antibody, a mouse anti- DDDK Tag antibody (AE005), a rabbit anti GFP-Tag pAb (AE011) from ABclonal, China, a rabbit anti SVCV-G pAb from Xueqin Liu (Huazhong Agricultural University, college of fisheries).The membrane was first washed for 5 × 6 min with TBS-T (TBS containing 0.1% Tween 20) solution, and then it was incubated for 45 min with the secondary antibody goat anti- mouse IgG (H1L) HRP (S0001, Affinity Biosciences) or anti-rabbit IgG HRP (S0001, Affinity Biosciences). The phosphorylation of STAT1a after recombinant CaIFNa1 or CaIFNυ induction was detected by primary antibody (Phospho-STAT1-Y701 Rabbit mAb (AP0054)) and secondary antibody goat anti-rabbit IgG HRP (S0001, Affinity Biosciences).
For fluorescence confocal, FHM cells were cultivated on a confocal dish at 1 × 105cells/well. Utilizing LipofectamineTM 3000 Transfection Reagent (Invitrogen, America), cells were transfected with the pEGFP-N1-STAT1a plasmid. Following a 12 h, the cells were subjected to 60 ng/ml of either CaIFNa1 or CaIFNυ recombinant proteins. After 12 h, the cells were washed with PBS and fixed for 10 min with 4% paraformaldehyde. Cell nuclei were stained with 4',6-Diamidino-2-phenylindole (DAPI) (Beyotime; C1006). It was then cleaned three times using PBS. The original photos were taken with an Olympus FluoView 1000 confocal microscope.
Co-immunoprecipitation
Following the manufacturer's instructions, the PierceTM Cross-linked Magnetic IP/Co-IP Kit (Thermo ScientificTM) was used to carry out the co- immunoprecipitation. In brief, HEK293T cells were seeded in 10-cm tissue culture dishes for 24 h and then co-transfected with 30 μg of pEGFP-N1-CaIFNυ and pcDNA4.0-CaCRFB4 and pcDNA3.1-Myc-CaCRFB12 (at a ratio of 1:1:1) using 90 μl of Lipofectamine™ 3000 Transfection Reagent (Invitrogen, America), and the three plasmids was co-converted. The cells were taken 48 h after transfection, and they were lysed for 30 min at 4 °C using cell lysis buffer (Beyotime, Nantong, China). Following that, the lysed cells were centrifuged for 30 min at 12,000 × g at 4°C. To eliminate background, 80 μl of lysate supernatant was mixed with 30 µl agarose resin. The corresponding immobilized primary antibodies were then added, and an overnight incubation at 4 °C was conducted using a Rotary mixer (MX-RD-Pro) (Dalong, China). The antibodies included Mouse anti GFP-Tag mAb (AE030, ABclonal, China), Rabbit anti Myc-Tag mAb (AE070, ABclonal, China), or Mouse anti His-Tag mAb (3040ES, Yeasen, China). Then, the mixed compound was centrifuged at 3,000 g for 5 min at 4 °C, 200 μl of lysis wash buffer was used to wash it three times, and the protein complexes sample was eluted using 100 μl of elution buffer. Subsequently, by adding 5 × SDS sample loading buffer for western blot analysis,12% SDS-PAGE was applied, and ECL system was used to visualize immunoreactive proteins.
Gene knockdown
siRNA was purchased from TSINGKE Biotechnology Ltd. (TSINGKE, China). The siRNA of CRFB12(XP_039517846.1) and CRFB4(XP_039506278.1) in Fimephales promelas were designed as CRFB12-siRNA-F: 5'-GGCUACGAGUAUGAACCAA-3', CRFB12-siRNA-R: 5'-UUGGUUCAUACUCGUAGCC-3'; CRFB4-siRNA-F: 5'-CGCUGGAGAUCUACGCUAA-3', 5'-UUAGCGUAGAUCUCCAGCG-3', Their knockdown effect was verified and shown in Figure S7-S8. Briefly, the FHM cells were seeded in a 6-well plate and transfected with siRNA of CRFB12 and CRFB4 using Lipofectamine™ 3000 Transfection Reagent (Invitrogen, America), according to the manufacturer’s protocol. Later, cells were co-transfected with STAT1a plasmids, recombinant CaIFNa1, or CaIFNυ induction for 12 h. Cells were collected for the transcriptional detection of jak, ifr9, stat1a, stat2, mx1, pkr, gig1, viperin gene, and the phosphorylation of STAT1a.
Statistical analysis
One or two-way ANOVA -test analysis was used in the statistical analysis, which was carried out with the Graphpad Prism 7.0 program. Significant difference is indicated for p values (* p < 0.05, ** p < 0.01, or *** p < 0.001).








Comments (0)