
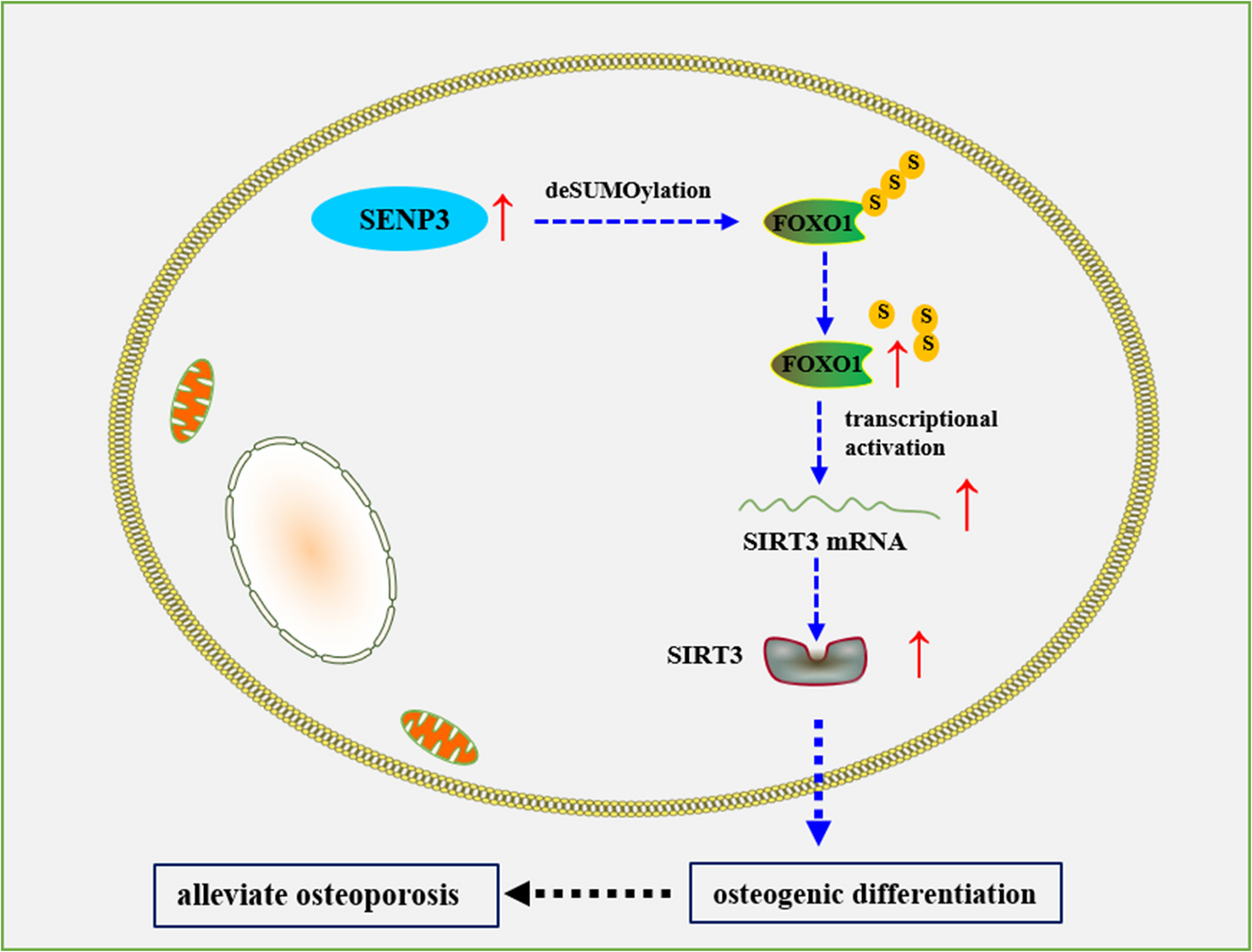
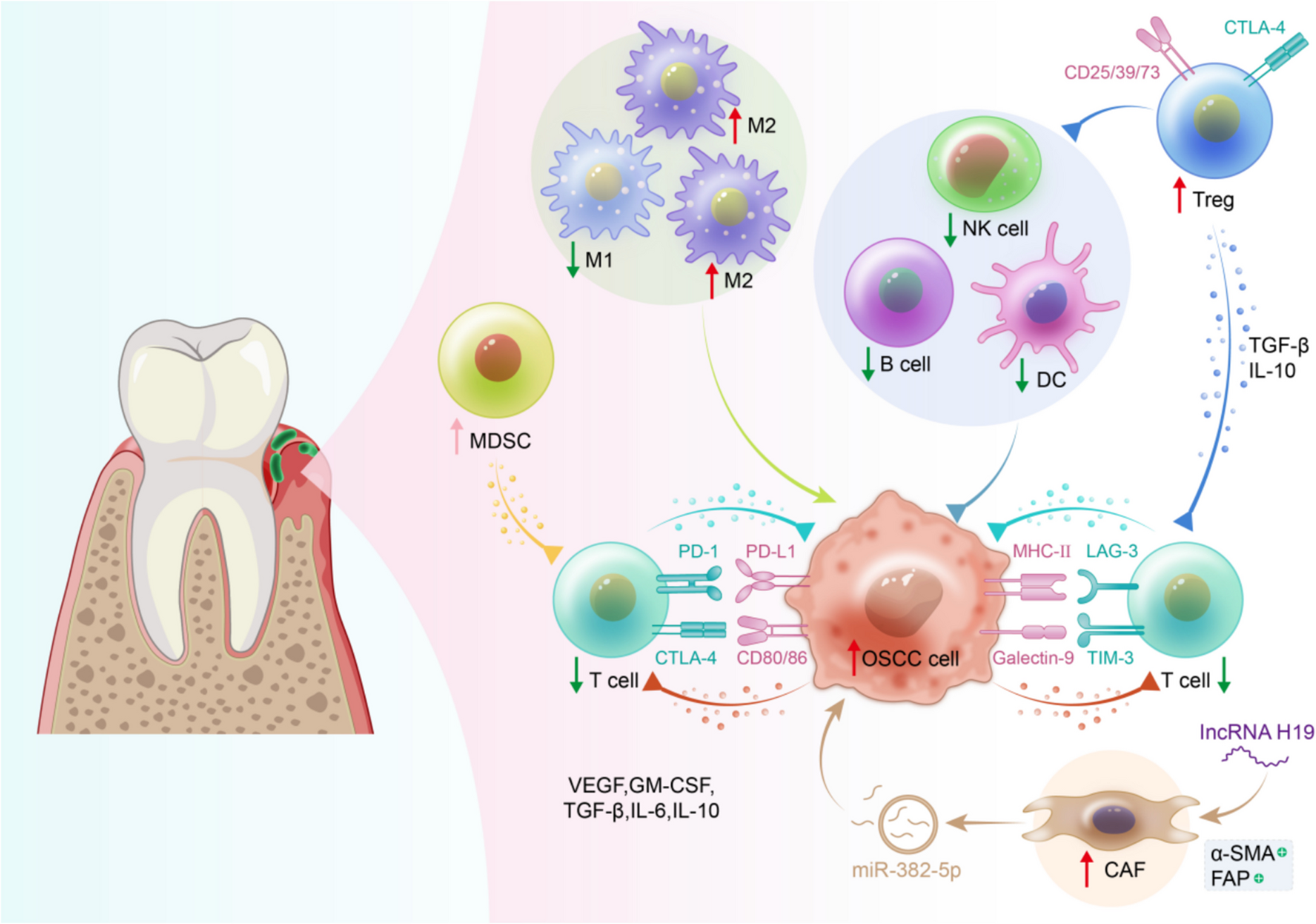
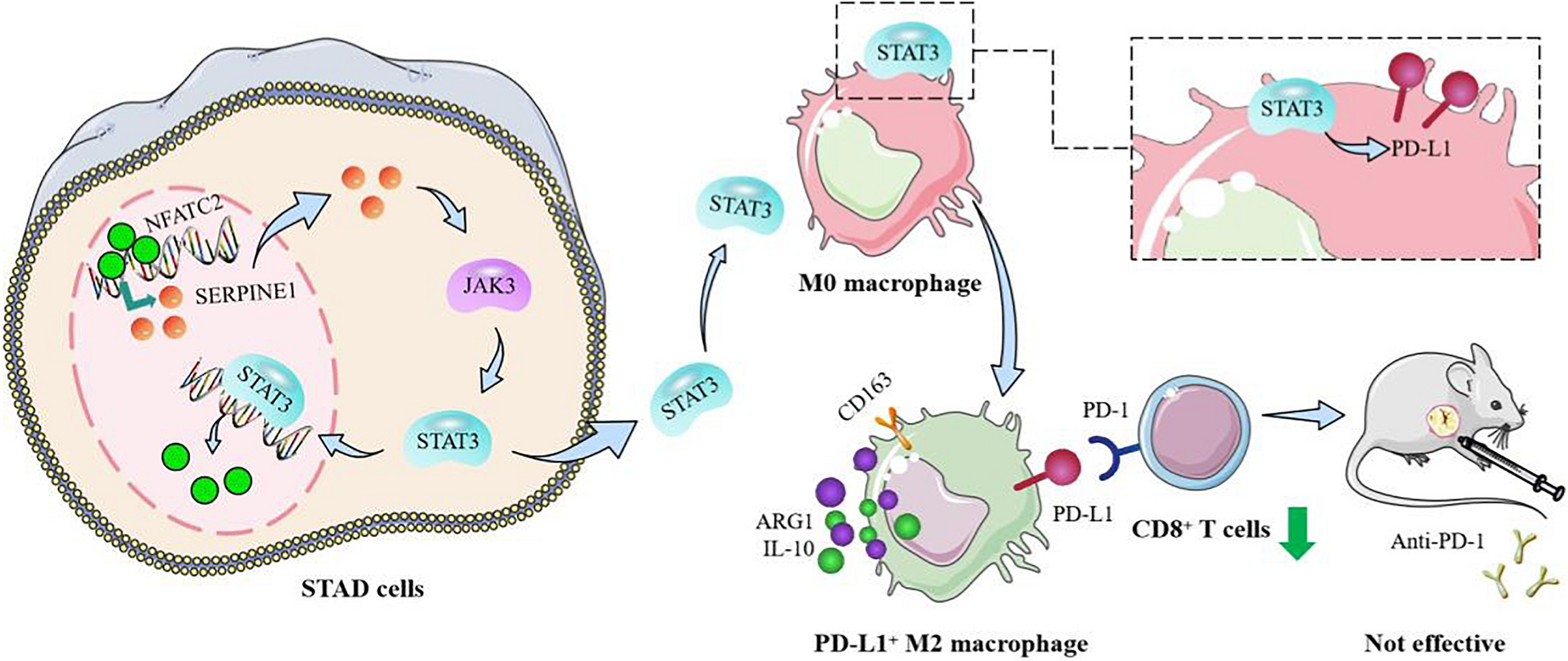
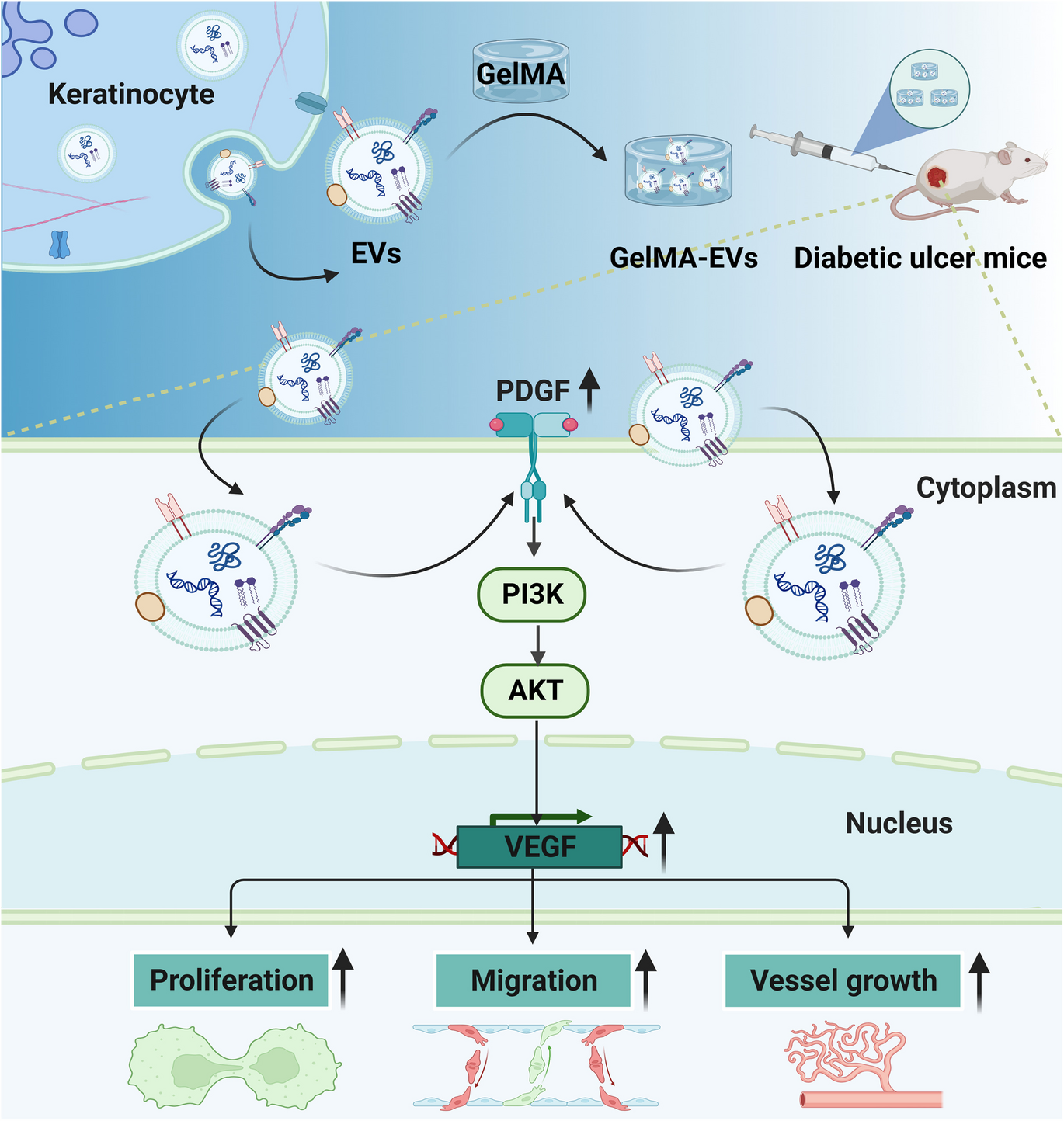
Remember me
We established a mouse model of paclitaxel-induced neuropathic pain according to the outlined procedure in Fig. 1A. We assessed mechanical allodynia, as well as thermal hyperalgesia and cold allodynia post-paclitaxel administration. As expected, a significant decrease in mechanical threshold was observed on day 3 post-injection, reaching the lowest levels on day 14 post-injection (Fig. 1B). Meanwhile, paw withdrawal latencies in response to thermal or cold stimulation declined on day 5 post-injection and were most pronouncedly affected on day 14 post-injection (Fig. 1C and D). These observations indicate that the repetitive administration of paclitaxel induced allodynia and hyperalgesia in mice.
Fig. 1
The development of a mouse model of paclitaxel-induced CIPN. A The process outline of development of a mouse model of paclitaxel-induced CIPN. B, C, and D Mechanical allodynia (B), thermal hyperalgesia (C) and cold allodynia (D) in mice following repeat administration of paclitaxel. The paw withdrawal thresholds in the von Frey test and response latencies in the hot and cold plate tests were measured before treatment (baseline) and after the first injection of either the vehicle or paclitaxel. The bar represents Mean ± SEM, with n = 6. The statistical significance is denoted as **p < 0.01, ***p < 0.001 vs. the naive group, and the statistical analysis was performed using two-way repeated-measures ANOVA with Tukey’s HSD correction
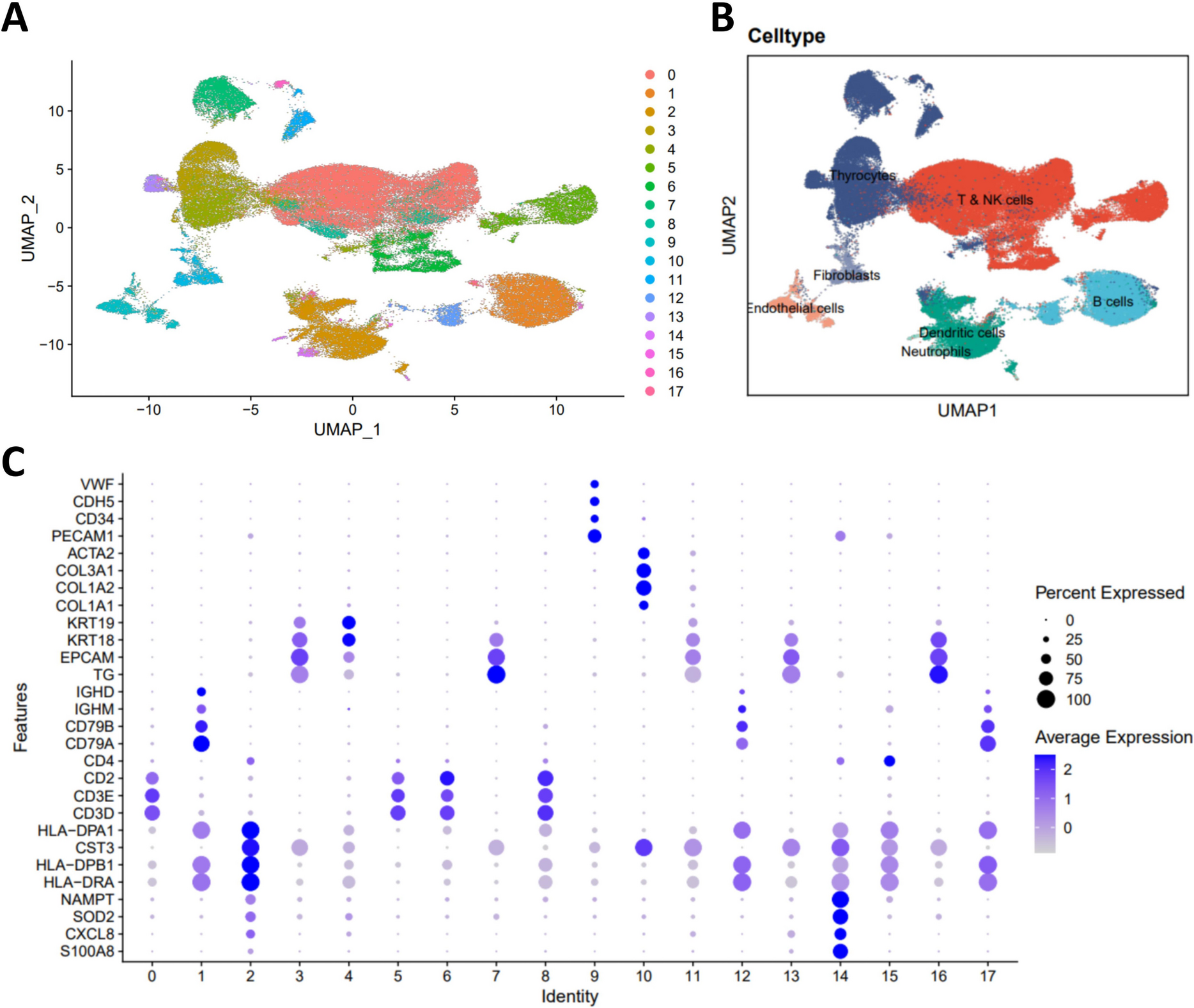
The neuronal and non-neuronal composition of DRGTo investigate cell type-specific pathogenesis in mice with paclitaxel-induced neuropathic pain, we conducted a single-nuclei RNA sequencing on lumbar (L) 4, 5, and 6 DRGs. The DRGs were isolated from mice (n = x/group) subjected to treatments involving saline for 14 days (referred to Ctrl), paclitaxel treatment for 14 days (referred to Pac14) and paclitaxel treatment followed by a recovery period of 7 days (referred to Pac21). The Fastq files were pre-processed and then mapped with the mouse mm10 reference genome to create expression matrix, cells and genes documents using MobiVision V3 software (MobiDrop). The processed data were loaded separately to the Seurat package (V5) and merged without integration. We filtered out cells exhibited mitochondrial genes percentage exceeding 5%, RNA counts fewer than 500, RNA counts over fivefold of the median RNA counts, and number of features exceeding fivefold of the median RNA features. The merged data underwent analysis using “LogNormalize” normalization and “pca” dimension reduction. Subsequently, the first 30 dimensions were input for constructing SNN group, with a resolution of 0.1 applied to differentiate various cell clusters. A total of 8 major cell populations were identified in the DRG (Fig. 2A), including Snap25 + neurons, Apoe + satellite glial cells (SGC), Mpz + Schwann cells (Schwann), Dcn + Fibroblasts (Fibro), Flt1 + endothelia cells (Endo), Myl1 + vascular smooth muscle cells (VSMC), and Ptprc + immune cells (Immune) (Fig. 2B-2F). One cluster could not be defined and categorized as undetermined (Und, possibly contaminated cells) due to a lack of known cell type-specific markers (Fig. 2B).
Fig. 2
The heterogeneity of dorsal root ganglia (DRG) cells in mice with paclitaxel-induced CIPN. A t-distributed scholastic neighbor embedding (t-SNE) plot of single cells profiled in the study, with each cell type represented by a colored dot. B The dot plot of the cell types in the DRG of paclitaxel-induced CIPN mice, marked by distinct genes. C, D, E, and F t-SNE plots of single cell types of Endo (C), VSMC (D), Fibro (E), and Schwann cells (F), respectively
The somatosensory neuron clusters in DRGWe then focused on the neuron cluster and utilized the list of DRG neuron markers summarized by Wang et al. (Wang et al. 2021b) to discern distinct neuronal populations within the DRG. We subset the counts of neuron cluster and perform a similar analysis as described above. The first 30 dimensions were used in PCA reduction and a resolution of 0.5 was set to distinguish neuronal subclusters. We noticed that two clusters (cluster 0 and 1) had very low RNA counts (counts of all cells < 1000) and lacked specific markers. These cells were considered low quality and removed by setting the threshold at nCount_RNA > 1000. The remaining data were then re-analyzed. We successfully identified 12 neuronal clusters, including Wnt7a + proprioceptors (Proprio/Wnt7a), Prokr2 + proprioceptors (Proprio/Prokr2), Ptgfr + Aβ low-threshold mechanoreceptors (Aβ_LTMR), Cadps2 + Aẟ_LTMR, Tafa4 + C_LTMR, Th +/Tafa4 + C_LTMR/Th, Sstr2 + thermal nociceptors (Heat), Rxfp1 + thermal nociceptors (Heat/Rxfp1), Trpm8 + cold nociceptors (Cold), Mrgprd +/Lpar3 + pruriceptors (Itch/Lpar3), Nppb + pruriceptors (Itch/Nppb) and Mrgpra3 + pruriceptors (Itch/Mrgpra3). A small cluster included multiple markers and was considered a mixed cell cluster. The t-SNE and UMAP coordinates of all neurons were plotted and demonstrated (Fig. 3A-3B). The dot plot of neuronal subtype-specific markers was also displayed (Fig. 3C).
Fig. 3
The heterogeneity of DRG neurons in mice with paclitaxel-induced CIPN. A and B t-SNE (A) and UMAP (B) plots of single cells profiled in the study, with each neuron type represented by a colored dot. C The dot plot of the neuron types in the DRG of paclitaxel-induced CIPN mice, marked by distinct genes
The gene ontology (GO) annotation of DEGs suggested potential alterations in nerve fiber and potassium-related currents in C_LTMR following paclitaxel treatmentWe then performed the analysis of differentially expressed genes (DEGs) across all neuronal subclusters between Ctrl and Pac14 or Pac21. The gene expression in the minimum percentage of cells (min.pct) and the average log2 fold change (avg_logFC) between groups were set at 0.25. The results unveiled notable up- and down-regulated DEGs across all neuron types within the DRG at both day 14 and 21 post-paclitaxel injection (Fig. 4A and B). Several genes, including Calcium/calmodulin dependent protein kinase 1D (Camk1d), 18S ribosomal RNA (Rn18s), and Cyclin dependent kinase 8 (Cdk8) were commonly up-regulated in multiple neuronal subtypes at two time points, indicating that these genes may be markers of paclitaxel exposure. Surprisingly, at the current sequencing depth, we were unable to quantified the expression of Atf3 gene (counts of all cell = 0), a marker of neuronal injury. It is worth noting that the C_LTMR and Itch/Lpar3 cluster had more DEGs compared to other clusters at both time points (Fig. 4A and B), suggesting that these two clusters were most vulnerable to paclitaxel treatment at the transcriptome levels. In this study, subsequent analysis focused on C_LTMR, which has been associated with pain hypersensitivity in many models of chronic pain.
Fig. 4
The transcripts regulated in all DRG neurons in mice with paclitaxel-induced CIPN. A and B The volcano plots showing the up- and down-regulated DEGs (differentially expressed genes) in all types of neurons in the DRG at both 14 (A) and 21 (B) days after paclitaxel injection. C and D The comparison of gene ontology (GO) enrichment of the up-regulated DEGs in C_LTMR neurons following paclitaxel injection at 14 (C) and 21 (D) days in mice. E and F The comparison of gene ontology (GO) enrichment of the down-regulated DEGs in C_LTMR neurons following paclitaxel injection at 14 (E) and 21 (F) days in mice. Top 10 significantly enriched GO terms, including biological process, cellular component, and molecular function, are shown
Gene ontology (GO) analysis of the DEGs in the C_LTMR cluster was then performed. Analysis of up-regulated DEGs at day 14 post-paclitaxel injection failed to enrich biological processes (BP) and molecular function (MF) terms, but highlighted the association of cellular component (CC) terms such as"mitochondrial ribosome","mitochondrial matrix", and"neuron projection terminus"(Fig. 4C), suggesting that mitochondrion and axonal fibers of C_LTMR might have been affected. At day 21 post-injection (Fig. 4D), enriched biological processes encompassed"RNA splicing","mRNA processing", and"signal transduction in response to DNA damage"in C_LTMR neurons. Enriched cellular component terms included"spliceosomal complex","neuron projection terminus", and"neurofilament", while enriched molecular functions were linked to"RNA helicase activity","pre-mRNA binding"and"calmodulin binding". These results indicated that there may be persistent impacts on axonal fibers of C_LTMR and reorganization of RNA processing during paclitaxel recovery.
For down-regulated DEGs, enriched cellular component terms such as"postsynaptic specialization","postsynaptic density","neuron to neuron synapse", and"potassium channel complex"were revealed at 14 days post-injection (Fig. 4E). At 21 days post-injection (Fig. 4F), enriched biological processes encompassed"synaptic vesicle cycle","cellular response to cAMP", and"regulation of membrane potential"in C_LTMR neurons. Additionally, enriched cellular component terms included"synaptic membrane","potassium channel complex","GABAergic synapse", and"asymmetric synapse", while enriched molecular functions were associated with"ion channel regulator activity","deacetylase activity", and"potassium channel activity". This analysis implies that paclitaxel treatment could lead to dysregulation in potassium related currents, which might subsequently affect the excitability of C_LTMR.
Gene set enrichment analysis (GSEA) of transcriptomic changes in C_LTMR neuronsWhile enrichment analysis using DEGs reveals prominent treatment-related ontologies, it may overlook pathways associated with DEGs exhibiting subthreshold fold changes. To address this limitation, we employed the GSEA method to identify affected Gene Ontology Biological Processes (GOBPs) in C_LTMR cluster. The Seurat object of neurons was loaded to escape (Easy single cell analysis platform for enrichment) package and subjected to GOBP enrichment analysis. As depicted in Fig. 5, aligning with the well-defined effects of paclitaxel treatment, the enrichment scores of several biological processes, including “microtubule-based transport”, and “oxidative phosphorylation,” and “mitochondrion organization” were significantly decreased in C_LTMR neurons treated with paclitaxel for 14 days compared to saline (Fig. 5A, B, C, and D). This result confirms the disruption of microtubule-related functions and reveals downstream mitochondrial deficits in C_LTMRs in CIPN mouse, providing a pathway-centric explanation for neurotoxicity.
Fig. 5
The gene set enrichment analysis of transcriptomic changes in C_LTMR neurons. A, B, C, and D The desregulated terms in C_LTMR neurons in mice, showing that paclitaxel treatment was associated with several biological processes, such as"long-term depression"(A),"microtubule-based transport"(B),"oxidative phosphorylation"(C), and"mitochondrion organization"(D). E The enrichment score of Interleukin 17 production in C_LTMR neurons in mice after paclitaxel treatment (E)
Pseudo-time analysis of C_LTMR clusterPseudo-time analysis enables the discovery of different cell states that exhibit subtle differences in cellular behaviours within a cell population. We subset the C_LTMR cluster and constructed pseudo-time trajectories to identify different cell states after paclitaxel treatment and genes with altered expression as the neurons transitioned between states (Fig. 6A). A total of 9 states were identified across the trajectories (Fig. 6B and C). Interestingly, the distribution of cell states varies among different groups, notably with a substantial increase in the prevalence of state 1 observed in two paclitaxel treatment groups. This result suggest that C_LTMR in state 1, positioned at the earliest point of the pseudo-time trajectory, represent a responsive state following paclitaxel exposure (Fig. 6B-D). We then performed unsupervised clustering analysis of gene expression and categorized genes according to their expression trends across the pseudo-time frame. This assigned genes into 6 different clusters. Notably, genes in cluster 2 and 4 exhibited a decreasing trend, whereas cluster 1 demonstrated an increasing pattern across the pseudo-time trajectory, indicating a close association with state 1 (Fig. 6E). The GO annotations of clustered genes show enrichment for various neurotransmission related terms, such as “synapse organization”, “axonogenesis”, “regulation of membrane potential” and “vesicle-mediated transport in synapse”. On the other hand, genes in clusters less likely associated with state 1 were more relevant to transcription modulation (e.g. “histone modification” and “chromatin “remodelling”). This result implies that the sustained presence of state 1 could directly contribute to abnormal pain processing after paclitaxel administration (Fig. 6E).
Fig. 6
Pseudotime analysis of transcriptomic changes in C_LTMR neurons. A Pseudo-time trajectories show the transition of the process in paclitaxel-induced CIPN mouse DRG C_LTMR neurons. The dots represent cells, and the colors indicate neuron clusters or pseudotime. B Genes in C_LTMR neuronal switch process are classified into 9 modules based on their expressing patterns. C The separation of Modules 1, 2, and 3. D The bar graph showing the frequency ratio of each module. E Heatmaps that display the DEGs module based on their dynamic expression characteristics, as shown in pseudotime analysis, in C_LTMR neurons in paclitaxel-induced CIPN mice. Gene ontology (GO) enrichment analysis results were used to analyze the biological processes of each gene module
Gene interaction analysis unveiled gene modules associated with altered biological processes in state 1 C_LTMRThe products of genes interact physically or co-occur to accomplish biological processes. To identify gene modules potentially involved in altered biological processes in state 1 C_LTMR, we extracted the genes from cluster 2 (Fig. 6E), which might have relevance to state 1, and constructed a gene interaction network using STRING (Fig. 7A). The MCODE plugin of Cytoscape software revealed numerous densely connected subnetworks, notably highlighting subnetwork 1, which could explain changes related to mitochondrion (Fig. 7B and C), and subnetwork 2, which could be relevant to alterations in neurotransmission (Fig. 7D and E). Additionally, the existence of subnetwork 3 also suggest that paclitaxel might dysregulate the ubiquitination system in C_LTMR cluster (Fig. 7F and G).
Fig. 7
Gene interaction analysis in state 1 C_LTMR neurons in mice with paclitaxel-induced CIPN. A Protein–protein interaction (PPI) network generated from genes in cluster 2, corresponding to pseudotime state 1. The network was constructed using STRING and analyzed with the MODE algorithm to identify functional modules. Nodes represent genes, and subnetworks identified as significant by MODE are shown in distinct colors, while non-significant subnetworks are displayed as purple nodes. B, D, and F The MCODE plugin of Cytoscape software revealed numerous densely connected subnetworks, notably highlighting subnetwork 1 (B), subnetwork 2 (D), and subnetwork 3 (F). C, E, and G GO analyses were conducted to determine the biological processes associated with each of these subnetworks 1 (C), subnetwork 2 (E), and subnetwork 3 (G) in state 1 C_LTMR neurons in mice with paclitaxel-induced CIPN
The up-regulation of camk1d contributed to thermal hyperalgesia and cold allodynia in mice with paclitaxel-induced CIPNCamk1d was commonly upregulated across multiple neuronal subtypes at two time points, suggesting its potential role as a biomarker of paclitaxel exposure. Feature plots (using UMAP dimensionality reduction) and violin plots illustrated this upregulation in paclitaxel-treated DRG neurons (Fig. 8A and B). This increase was confirmed by in situ hybridization in the DRG of mice with paclitaxel-induced CIPN, which demonstrated upregulation in multiple neuronal subtypes at both time points (Fig. 8C and D). To further investigate the function of Camk1d in CIPN, we performed Camk1d knockdown in the DRG of CIPN mice (Fig. 8E). The results reveal that mechanical allodynia (increased sensitivity to touch) was unaffected by Camk1d knockdown (Fig. 8F). However, Camk1d knockdown significantly reduced thermal hyperalgesia (increased sensitivity to heat) and cold allodynia (increased sensitivity to cold) in response to thermal or cold stimuli (Fig. 8G and H). These findings indicate that Camk1d contributes to temperature hypersensitivity, a key clinical symptom of CIPN in humans. In contrast, Camk1d does not appear to mediate mechanical allodynia in this mouse model of CIPN.
Fig. 8
The expression and function of Camk1d in DRG in mice with paclitaxel-induced CIPN. A Feature plots of Camk1d expression using UMAP for dimensionality reduction. B Violin plots illustrating its upregulation following paclitaxel treatment. C In situ hybridization images exhibit the expression of Camk1d (green) and DAPI (blue) in DRG in mice with 0, 14, and 21-day paclitaxel-induced CIPN. D The quantification of the fluorescence signal from panel A. E The mRNA expression of Camk1d in DRG in mice after Camk1d knockdown. F, G, and H Mechanical allodynia (F), thermal hyperalgesia (G) and cold allodynia (H) in mice with or without knockdown of Camk1d following repeated administration of paclitaxel. The paw withdrawal thresholds in the von Frey test and response latencies in the hot and cold plate tests were measured before treatment (baseline) and after the first injection of either the vehicle or paclitaxel. Mean ± SEM, n = 6. The statistical significance is denoted as **p < 0.01, ***p < 0.001 vs. the GFP + saline group, #p < 0.05, ##p < 0.01 vs. the PTX + Camk1d group and the statistical analysis was performed using two-way repeated-measures ANOVA with Tukey’s HSD correction
Comments (0)