
Remember me




Hyponatremic hypertensive syndrome was first reported in adults, and is a rare condition in children. It is typically characterized by hypertension and hyponatremia and can be caused by a number of disorders including kidney failure, malignant hypertension, thiazide use, renin-secreting tumors and renal ischemia [1]. Hyponatremic hypertensive syndrome caused by renal ischemia is rare [2]. The commonest etiology in children is unilateral renal artery stenosis. Early diagnosis and timely treatment are crucial to avoid complications such as posterior reversible encephalopathy syndrome, which may arise due to uncontrolled hypertension combined with cerebral edema from hyponatremia [3]. Hyponatremic hypertensive syndrome usually presents with neurological symptoms such as headache, confusion, and seizures, along with weight loss, polydipsia, and polyuria. Laboratory findings include hyponatremia, hyperreninemia, hypokalemia, hyperaldosteronism, metabolic alkalosis, and increased urinary sodium and protein excretion [1].
Renovascular hypertension should be considered in children with suspected secondary hypertension, particularly when associated with elevated plasma renin, hypokalemia, or severe hypertension requiring more than two antihypertensive agents to be controlled [4]. A number of causes of renovascular disease are shown in Table 1S (supplemental material).
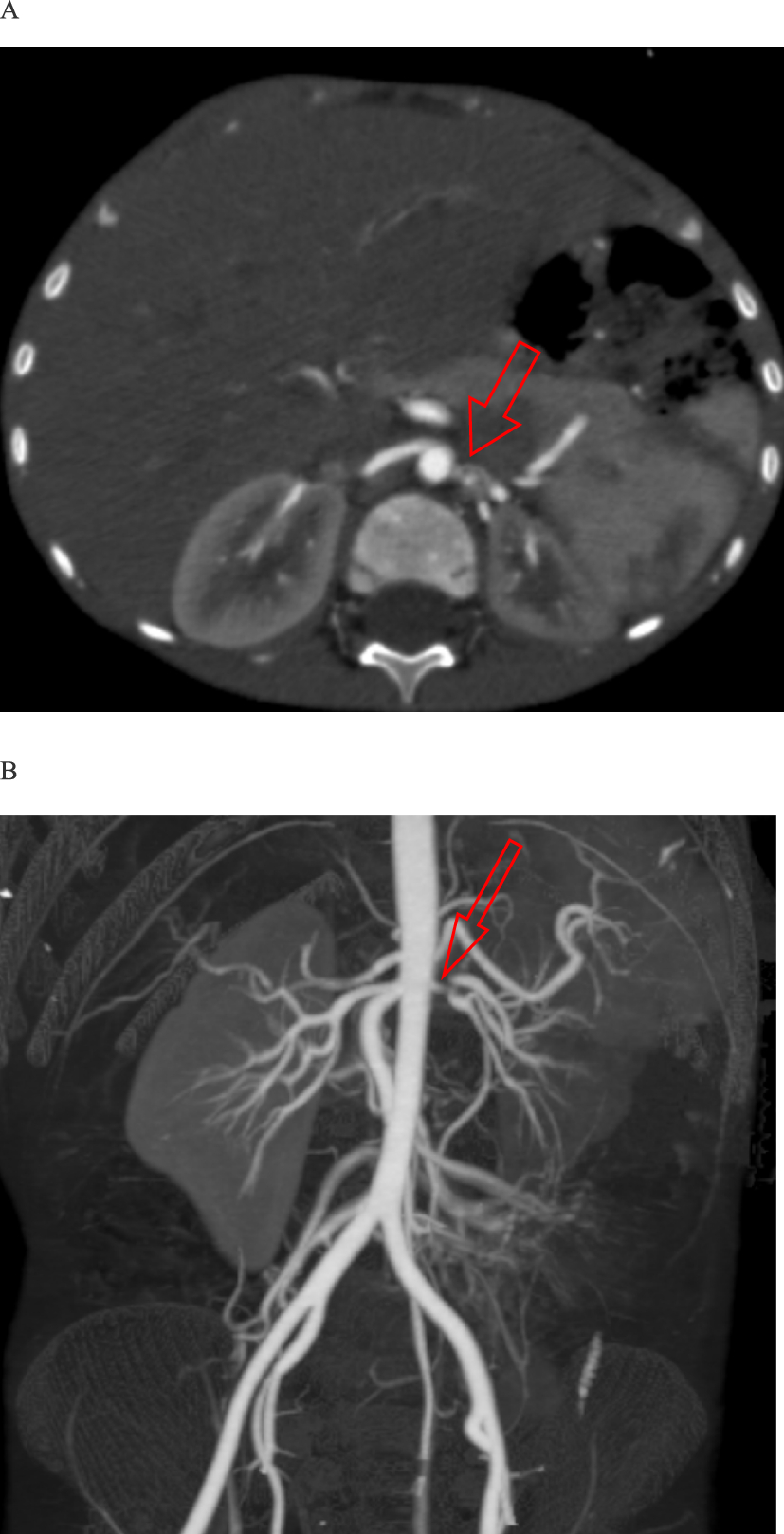
This is a case of 5-year-old male who presented with seizures and severe hypertension. Coarctation of the aorta was unlikely due to the absence of blood pressure discrepancies between the upper and lower limbs, which was further confirmed by echocardiography. There were no signs of systemic or renal parenchymal disease. The presentation with hypertension, hyponatremia, hypokalemia, and polyuria, strongly suggested hyponatremic hypertensive syndrome. Brain MRI findings were consistent with posterior reversible encephalopathy syndrome.
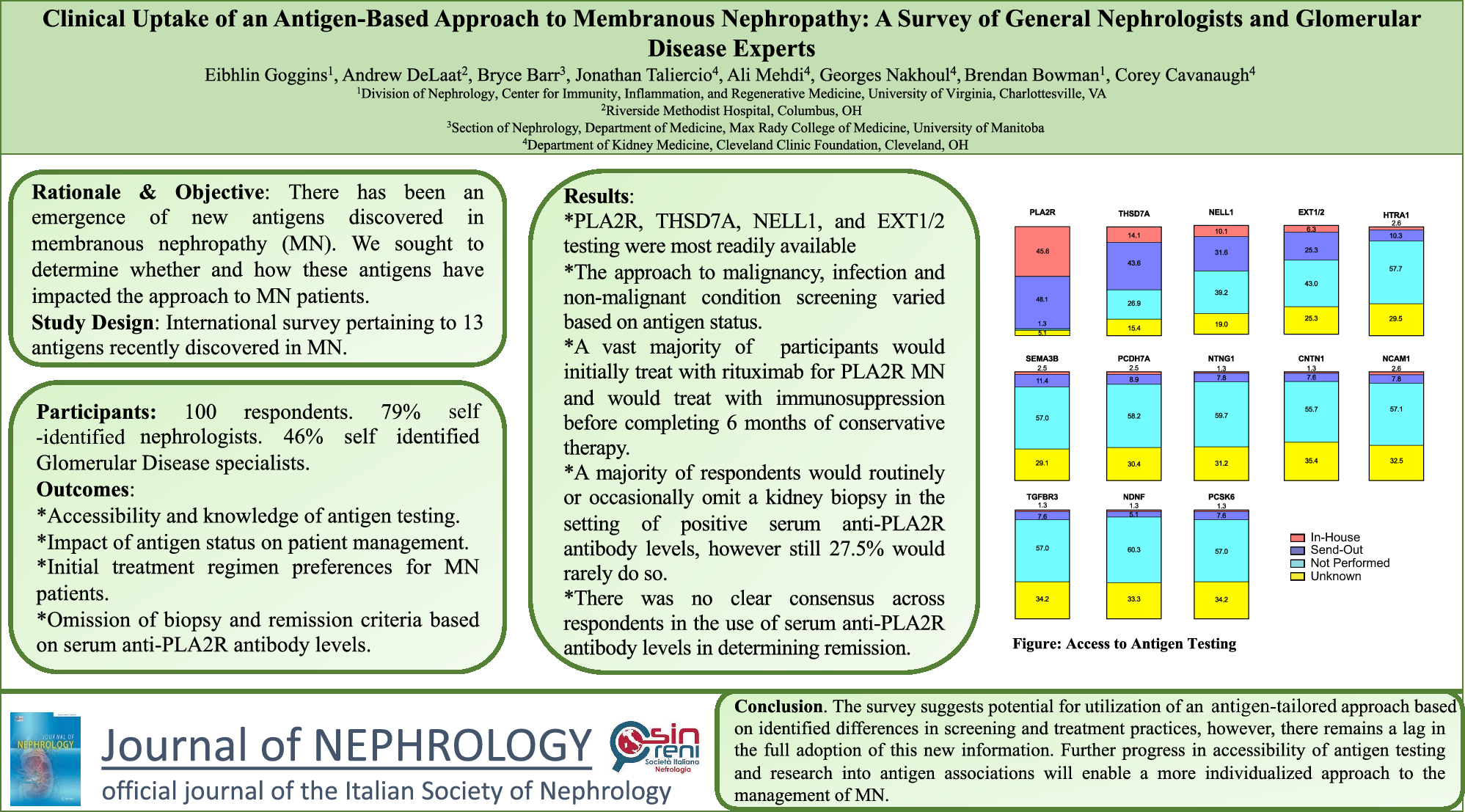
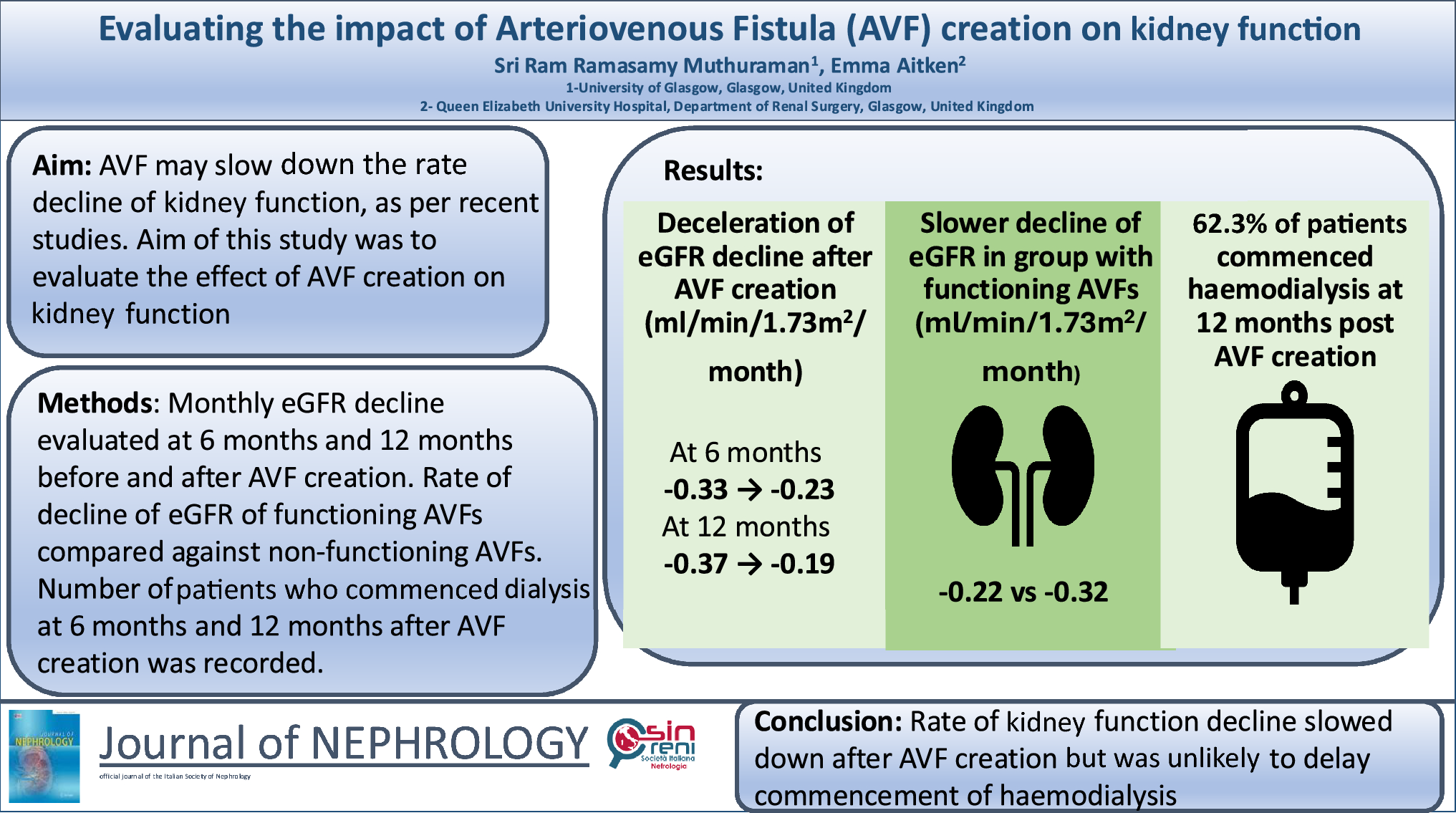
The main pathogenesis of hyponatremic hypertensive syndrome is renal ischemia, as shown in Fig. 2 [3]. Renal ischemia leads to increased renin secretion, which elevates angiotensin II levels. This, in turn, raises blood pressure through direct vasoconstriction and secondary hyperaldosteronism. A rapid rise in arterial pressure can cause glomerular hyperfiltration and pressure natriuresis in the non-stenotic kidney, causing volume depletion and hyponatremia, which may result in further renin release from the ischemic kidney [5]. Hypokalemia, resulting from hyperaldosteronism, exacerbates renin secretion, thus intensifying the vicious circle. Additionally, hyponatremia can be further exacerbated by stimulation of thirst and antidiuretic hormone release, triggered by both high angiotensin II levels and volume depletion. Metabolic alkalosis is also a consequence of renin–angiotensin–aldosterone system activation [3]. Glomerular hyperfiltration in the contralateral healthy kidney may lead to tubulointerstitial damage by hypercalciuria and hyperuricosuria. Proteinuria in hyponatremic hypertensive syndrome can result from glomerular hyperfiltration, the proteinuric effects of angiotensin II, or tubulointerstitial injury from hypercalciuria and hyperuricosuria [3].
Fig. 2
Possible mechanism of hyponatremic-hypertensive syndrome [3]
Ding et al. [3] reviewed 15 pediatric cases of hyponatremic hypertensive syndrome secondary to unilateral renal artery stenosis, reporting a mean age of onset of 4.03 ± 3.38 years and a male predominance (11 out of 15), consistent with the age and gender of our patient. The most common clinical features were hypertension, polydipsia, and polyuria (14/15), followed by hyponatremic seizures (7/15). Similarly, Agarwal et al. [2] described 32 adult cases, with predominant symptoms including headache, altered consciousness, or confusion (24/32), and features such as weakness, weight loss, thirst, and polyuria (15/32). Papilledema or retinal hemorrhage was seen in 7 patients. Our patient had comparable findings, with hypertensive retinopathy observed on fundus examination of the left eye.
An important differential diagnosis for this presentation includes causes of hypokalemic hypertension. Liddle syndrome, resulting from mutations in the epithelial sodium channel, leads to aldosterone-independent sodium reabsorption in the distal nephron. It typically presents with hypertension, polyuria, hypokalemia, and metabolic alkalosis, along with suppressed plasma renin activity and low aldosterone levels, often accompanied by hypernatremia. Similarly, apparent mineralocorticoid excess, an autosomal recessive disorder caused by mutations in the HSD11B2 gene encoding 11β-hydroxysteroid dehydrogenase type 2, presents with juvenile-onset severe hypertension, low renin and aldosterone, hypokalemia, and metabolic alkalosis. Familial hyperaldosteronism also leads to hypokalemic hypertension and metabolic alkalosis, typically associated with inappropriately elevated aldosterone levels and hypernatremia.
The management of hyponatremic hypertensive syndrome secondary to renal artery stenosis involves correction of volume depletion and hypokalemia, blood pressure control, and definitive treatment of the underlying renal artery stenosis. Restoring intravascular volume is a priority to improve systemic perfusion and minimize further renal ischemic injury [6]. Hypertension can be controlled by administering intravenous calcium channel blockers, while diuretics are avoided to prevent exacerbation of fluid and sodium loss and further stimulation of the renin–angiotensin–aldosterone system [7]. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers should be introduced in patients with hyponatremic hypertensive syndrome to block the over-activation of the renin–angiotensin–aldosterone system. However, they are generally contraindicated as first line antihypertensives, particularly in bilateral disease or in patients with a solitary kidney [4]. Surgical correction of renal artery stenosis can be achieved by renal angioplasty, renal artery reconstruction or unilateral nephrectomy, with the latter considered when the affected kidney contributes less than 10% to total kidney function or if percutaneous angioplasty fails [8]. In our case, the patient was initially stabilized with intravenous hydralazine, beta-blockers, captopril and calcium channel blockers, followed by successful management with left nephrectomy.
Delayed recognition and inadequate treatment of hyponatremic hypertensive syndrome can result in irreversible renal ischemia, end-organ damage, such as left ventricular hypertrophy, and neurological injury from hypertensive or hyponatremic encephalopathy [6]. The prognosis for posterior reversible encephalopathy syndrome associated with renal disease is generally favorable, with most patients recovering without lasting neurological deficits [9].
In conclusion, this rare case of unilateral renal artery stenosis in a young child emphasizes the importance of early recognition and appropriate management. This report contributes to the limited pediatric literature on hyponatremic hypertensive syndrome and advocates for heightened awareness among pediatricians managing hypertension.
Comments (0)