
Remember me
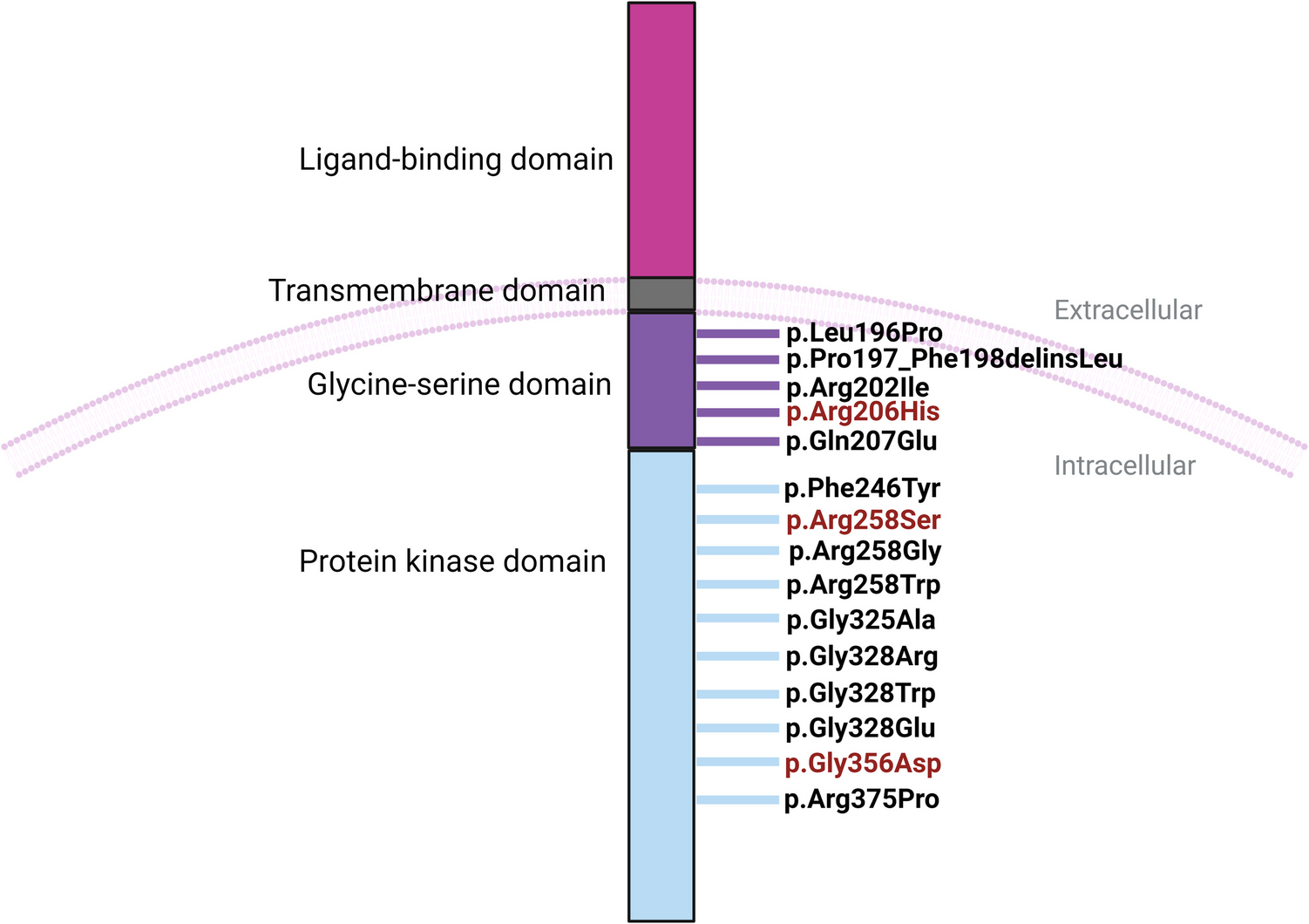

The patient is a 14-year-old Caucasian female with confirmed TROFAS. She measured 161 cm and weighed 60 kg (both corresponding to approximately P55/+1.2 standard deviation score (SDS) and P70/+1.5 SDS for her age, respectively) and a BMI of 23.15. The patient is the second child of non-consanguineous parents, and the brother appears healthy. During the pregnancy, reduced fetal movements were noted and a spontaneous vaginal delivery occurred at 36th+4 weeks of gestation. At birth, the infant had a weight of 2420 gs (25th–50th percentile), a length of 49 cm (50th–75th percentile), and a head circumference of 32 cm (10th–25th percentile). Apgar scores were 8 at 1 min, 9 at 5 min, and 10 at 10 min (Fig. 1).
Developmental milestones were delayed, with independent walking achieved at approximately 2 years of age and delayed speech development noted. She began school at the age of 7 and required the support of a caregiver during the first year of middle school. According to the end-of-year school report, her cognitive abilities and academic skills were not age-appropriate. She required consistent routines in daily school activities and had difficulty adapting to changes or new demands. Notable challenges included reduced memory, increased distractibility, and impaired concentration. Additionally, she struggled with handling money, navigating traffic, and appropriately assessing dangerous situations.
At the age of six, the patient underwent to some genetic tests. Chromosomal analysis revealed a normal female karyotype: 46,XX (based on the evaluation of 50 metaphases). Interphase fluorescent in situ hybridization on buccal mucosa cells was performed to investigate a possible Turner mosaicism, with unremarkable findings. FISH analysis was also performed from the peripheral blood to rule out a microdeletion in the chromosomal region 22q11.2, using the DiGeorge/VCFS TUPLE1 Region Probe (Cytocell), with the karyotype reported as ish 22q11.2(HIRAx2). However, in all examined metaphases, both chromosome 22 showed specific hybridization signals for the HIRA (TUPLE1) gene as well as the control signals. Therefore, a microdeletion in the investigated chromosomal region could be ruled out with high probability. After several years without a definitive diagnosis, exome analysis at the age of 13 identified two heterozygous mutations in the FIBP gene: c.1003C>T, p.(Arg335*), and c.497_498del, p.(Ser166*). Parental testing revealed that each parent carries one of these mutations: the mother is heterozygous for the c.497_498del mutation, while the father is heterozygous for the c.1003C>T mutation. This confirms that the patient has inherited one mutated allele from each parent, resulting in a compound heterozygous state for the FIBP gene.
Fig. 2
Tonal audiometry showing a fluctuating transmission hearing loss due to ear ventilatory dysfunction
Pulmonary complications have been a significant issue, as the patient experienced recurrent respiratory infections, including bronchitis and pneumonia, attributed to a hypoplastic pulmonary artery. In July 2024, a bronchoscopy revealed a 50% obstruction of the main bronchus, leaving the left lung nonfunctional. Lung fibrosis was also noted, which further complicated her respiratory status.
The patient’s otolaryngological history is notable for subglottic tracheal stenosis, which has significantly contributed to her respiratory challenges. Since early childhood, she has experienced intermittent conductive hearing loss due to tubal dysfunction, without any evidence of sensorineural impairment. Figure 2 shows two tonal audiometry performed 2 months apart in which the right (red lines) and left (blue lines) transmission hearing loss changes at the level of aerial auditory perception (bottom line), which is due to tubal and thus ventilatory dysfunction.
Endoscopically, she presents bilateral hypertrophy of the Torus tubarius and grade III tonsillar hypertrophy (tonsils are beyond the pillars), despite undergoing an adeno-tonsillotomy in 2015 to alleviate obstructive symptoms and recurrent infections. Her tympanic membranes remain intact and free of sclerosis. Notably, she does not exhibit signs of sleep apnea. Dental evaluation has revealed a posterior crossbite malocclusion.
Her cardiac history is notable for congenital heart anomalies, including a double-chambered right ventricle, a patent foramen ovale, and a ventricular septal defect, all surgically repaired in January 2011. Additionally, the patient has left pulmonary vein atresia and a progressively enlarging mediastinal vascular conglomerate, predominantly peritracheal and left peribronchial, which has been documented since 2018. In June 2024, she developed a complete atrioventricular block, necessitating the implantation of a pacemaker in July 2024.
The patient also presents significant ophthalmologic findings. She was diagnosed with intermediate uveitis, for which she began Adalimumab therapy in December 2023. By October 2023, she developed increasing vitritis with snowballs in the left eye, while a known arterial anomaly in the right eye has remained stable (Fig. 3). Other causes of uveitis and AV block were excluded.
Fig. 3
Left: vitritis with snowballs in the left eye. Right: arterial anomaly
Additionally, a significant vitamin D deficiency was identified, warranting rheumatologic evaluation and supplementation. At birth, the patient presented with a left clubfoot deformity, which was managed conservatively with the redressive casts and a tenotomy of the Achilles tendon, according with Ponseti method (Heck et al. 2016).
In November 2024, she reported bilateral knee pain, and radiographic evaluation revealed benign, non-ossifying fibromas in both knees (Fig. 4), a finding not previously associated with TROFAS. Radiographical findings of the upper and lower extremities show the bowing of both sides of the tibia, radius and ulna, probably associated with low levels of vitamin D (Fig. 5).
Fig. 4
RX with arrows show the non-ossifying fibromas of the left and right knee
Fig. 5
RX of arm and leg show the bowing of radius, ulna, tibia, and fibula
Comments (0)