
Remember me
Mucopolysaccharidosis (MPS) III A, also known as Sanfilippo Syndrome type A (OMIM #252900), is an autosomal recessive disorder affecting the central nervous system (CNS). It is caused by biallelic pathogenic variants in the SGSH gene that lead to a deficiency in lysosomal heparan sulfamidase enzyme, required for degradation of heparan sulphate (HS) glycosaminoglycan. Lysosomal HS accumulation is associated with prominent neurological regression (progressive cognitive impairment, behavioral changes, epilepsy and sleep disturbances) and variable multisystem involvement with possible craniofacial dysmorphism, macrocephaly, dysostosis multiplex and valvular cardiopathy [1]. Behavioral phenotype of MPS III is considered a hallmark of the disease and one of the most challenging aspects of the disorder [2]. It is likely to be related to neurodegeneration resulting from HS accumulation with dysregulation of cell signaling, mitochondrial dysfunction, enhanced oxidative stress and impaired autophagy. These processes lead to neuroinflammation and defective neurotransmission [3]. Unlike other MPS, no disease-modifying therapy has yet been approved. Depending on the residual enzyme activity, MPS III patients more frequently present a severe phenotype (Rapid Progressing, RP) with onset in early childhood or, more rarely, a milder, attenuated phenotype (Slow Progressing, SP) with later onset, even in adulthood, and a slower regression of intellectual and motor abilities [4]. Patients with attenuated phenotype may present behavioral and psychiatric symptoms in the early stages of the disease, receiving various psychiatric diagnoses, such as Attention Deficit Hyperactivity Disorder, Oppositional Defiant Disorder, Autism Spectrum Disorder, until frank cognitive decline, usually in their late teens, prompts a laboratory diagnostic work-up [5].
We report a 16-year-old patient diagnosed with MPS III A after the onset of an acute and transient psychotic disorder (ATPD) (ICD-10, F23). The patient was born at term to non-consanguineous parents. Family history was remarkable for psychiatric disorders. His older brother suffers from an unspecified mood disorder, which is being managed with lithium and his mother has a history of migraines and an unspecified anxiety disorder. According to the parents’ annotations, the boy exhibited no cognitive or verbal impairment prior to the onset of the disease, and psychomotor development was referred to as normal. The parents reported that their son had exhibited symptoms of anxiety and mood swings during the SARS-CoV-2 pandemic. These psychological challenges were subsequently followed by academic difficulties and impairment in social interaction upon his return to school.
Two months prior to hospital admission, at the age of 15, the patient showed significant psychological distress at school, characterised by learning difficulties, sleep disturbances, headaches, fatigue and impaired attention skills. As a consequence, the primary care physician prescribed a range of supplements containing melatonin, valerian, magnesium, coenzyme Q10, vitamin B2, phosphoserine, glutamine, asparagine, and vitamin B6. This dietary intervention was employed as a generic approach to address the symptoms of mental and physical fatigue.
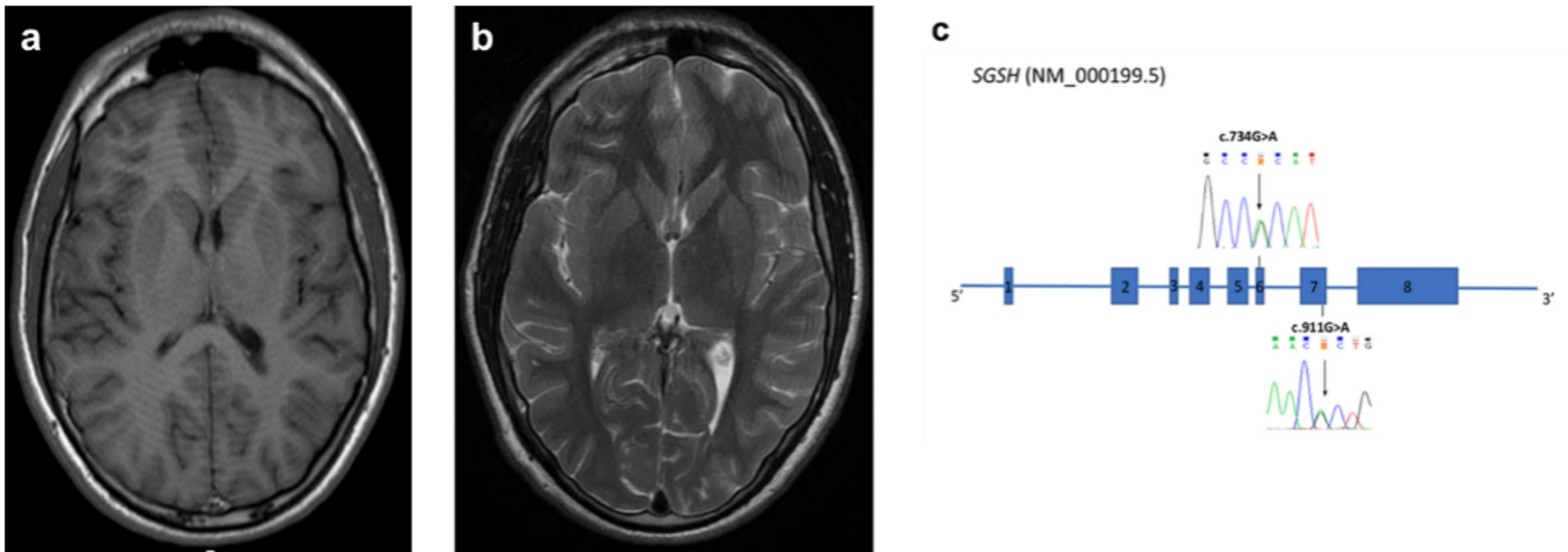
Due to the persistence of these symptoms and the onset of asthenopia, mutism, bradykinesia, social withdrawal, and spatial-temporal disorientation, he was admitted to the Pediatric Emergency Room and later transferred to the Pediatric Unit. Growth parameters were within the normal range; weight 67 kg (50-75th p.le), height 168 cm (25th p.le) and head circumference 56 cm (25-50th p.le). On physical examination conjunctival icterus and mild liver enlargement were reported. Routine blood analyses showed unconjugated hyperbilirubinemia (7–10 mg/dl; n.v. 0.3–1.2). Direct and indirect Coombs tests, blood ceruloplasmin levels and urinary copper excretion were normal. To identify specific etiological factors underlying psychotic symptoms, extensive serum and cerebrospinal fluid (CSF) investigations were performed ruling out inflammatory and infectious diseases of the CNS. Serum and CSF auto-antibodies analyses yielded normal results. Abdominal ultrasound examination showed hyperechogenic, enlarged liver. Electroencephalogram recording was unremarkable. Brain magnetic resonance imaging (MRI) showed cortical atrophy with enlargement of fronto-temporo-parietal subarachnoid spaces bilaterally (Fig. 1a). Intravenous immunoglobulin therapy was administered for a five-day period, yet no discernible benefit was observed. The patient was managed with Risperidone (1–2 mg/day), which was discontinued due to the onset of extrapyramidal signs. He was referred to our Adolescent Neuropsychiatry Unit for persistent behavioral problems. Neurological examination was remarkable with social and communicative impairment, disorganized speech with tendency to mutism, bradykinesia and ideo-motor apraxia. Aripiprazole therapy (2,5–15 mg/day) was established until complete remission of psychotic symptoms. After the apparent resolution of the clinical picture following eight weeks of treatment with Aripiprazole, the patient experienced additional recurrent transient psychotic episodes accompanied by regression of verbal language with progressive dysphasia, social anxiety and social withdrawal. At age 17 psychometric evaluation indicated mild intellectual disability (WISC-IV total IQ: 68) and mildly impaired adaptive level on the Vineland-II scale. Routine blood analyses showed mild, non-haemolytic, unconjugated hyperbilirubinemia. At the most recent evaluation (age 18), we observed a worsening of social withdrawal, and progressive lost of language organization. The patient is currently being treated with brexpiprazole (2–1 mg), which was started following an acute transient psychotic episode preceded by the occurrence of hyperbilirubinemia.
Genetic tests confirmed that the patient had Gilbert’s syndrome caused by homozygous variant (-41_-40dupTA)s in the TATAbox regions of the UGT1A1 gene (NM_000463).
Owing to the history of formal regression at school associated with behavioral and emotional challenges, we performed neurometabolic screening that showed repeated increased urinary mucopolysaccharides levels (7.60–9.80 mg/mmol, normal range 0.51–2.57). A whole exome sequencing was performed (4basesWholExProv3) and a virtual panel of genes associated with lysosomal storage diseases was analyzed. Two missense variants were identified and further confirmed by Sanger sequencing in the SGSH gene (NM_000199.5): c.734G > A p.(Arg245His) and c.911G > A p.(Arg304His) (Fig. 1c). The c.734G > A p.(Arg245His) variant was classified as pathogenic according to the guidelines of the American College of Medical Genetics (ACMG) (criteria: PP5, PM5, PP3, PM2, PP2). The variant was previously reported in patients affected by Sanfilippo IIIA and found to be related to the development of a severe phenotype [4, 6]. The c.911G > A p.(Arg304His) is a previously unreported variant, classified as a variant of unknown significance (VUS) according to ACMG guidelines (criteria: PP3, PM2, PP2). However, the analysis of the patient’s parents showed that the two SGSH identified variants were in trans. Based on this result, criteria PM3 was activated for the classification of the c.911G > A (variant identified in compound heterozygosis with a pathogenic variant), which was therefore re-classified as probably pathogenic. The diagnosis of MPS IIIA was confirmed by reduced SGSH enzyme activity in leukocytes (0.4 nmol/mg/17 h, normal range 4.3 ± 2.8). Repeated brain MRI showed a mild progression of cortical atrophy with increased enlargement of the supratentorial subarachnoid spaces (Fig. 1b).
Fig. 1
a-b Brain MRI of the study patient with SP MPS III A. (a) T1 axial sequence at the age of 16 years showing cortical atrophy with enlargement of temporo-parietal subarachnoid spaces. (b) T2 axial sequence at the age of 17 years showing a mild progression of cortical atrophy with increased enlargement of the supratentorial subarachnoid spaces. (c) Schematic representation of the SGSH gene (NM_000199.5) and the Sanger sequencing confirmation of the two variants, c.734G > A p.(Arg254His) and c.911G > A p.(Arg304His), identified by exon sequencing in exon 6 and 7 respectively
The present patient showed an acute psychiatric onset of MPS III A in adolescence characterized by rapid behavioral changes between states of stupor and restless agitation and disrupted sleep patterns preceded by headache, fatigue and attention deficit and consistent with a diagnosis of acute and transient psychotic disorder (ATPD) (ICD-10, F23). Later on, the patient had recurrent psychotic episodes responsive to antipsychotic drug therapy. Over the follow-up period (3 years) the subject exhibited a mild yet progressive cognitive decline, characterised by a deterioration in verbal communication skills and an escalation in social withdrawal and social anxiety.
Noteworthy, in the study patient developmental trajectory was normal with slow progression into mild intellectual disability, as previously described in some patients with SP MPS III A (attenuated phenotype) [7–8]. In particular, the abrupt onset of psychiatric symptoms in adolescence, suggestive of schizophreniform disorder, was reported in an 18-year-old patient with MPS III B who had an unremarkable psychomotor development and who presented with attention deficit and learning disabilities until the onset of acute psychiatric symptomatology [9]. In the present patient repeated brain MRI showed mild progression of cerebral atrophy with greater involvement of the fronto-temporo-parietal lobes. Notably, patients with SP MPS III A may present an attenuated decline of cortical grey matter volumes and little or no change of white matter volumes compared to patients with RP disease [1].
The heterogeneous phenotype of MPS III A and the variability in disease progression appear related to allelic heterogeneity with notable genotype-phenotype association [4]. Milder phenotype was described in patients compound heterozygous for a variant associated with an attenuated course of the disease (e.g. p.S298P, p.G122R, p.R206P) or harboring a previously unreported variant in combination with one of the severe mutations (e.g. p.R245H, p.Q380R, p.S66W) [4, 6]. In this context, further studies could confirm in patients with attenuated MPS IIIA the presence of the novel missense variant (p.R304H) which we found in combination with the severe mutation (p.R245H).
In conclusion, this study emphasizes the need for neurometabolic evaluation in adolescent patients with acute onset psychiatric symptoms, even in the absence of obvious neurological regression. Although biological bases underlying the neurobehavioral phenotype in individuals with MPS III have been extensively studied [3], the complex relationship between environmental, biological and familial factors and the effect of triggers might be considered for understanding differences among individual patients. Careful neurological and psychiatric assessment may enable an earlier diagnosis and a stratification of disease progression in MPS IIIA, particularly in patients with an attenuated course of the disease showing mild yet progressive cognitive decline prompting development of therapeutic approaches for early treatment [10].
Comments (0)