
Remember me
To assess the feasibility of conducting a larger randomized clinical trial investigating the Monarch eTNS device versus sham for four weeks in patients with ADHD in Denmark.
Trial designParallel-group, sham-controlled, feasibility randomised clinical trial.
Trial settingThe trial will be conducted at two outpatient child and adolescent psychiatric clinics, i.e. the Region of Southern Denmark (2 sites) and in Region Zealand (1 site).
Staff qualifications, and trainingThe investigators consist of psychiatrists and psychologists with expertise in child and adolescent psychiatry, and a neuroscientist with expertise in neuromodulation techniques. All investigators will be trained to use the device properly. Dedicated investigators will be responsible for instructing the families. Software and electrical engineers with expertise in medical devices will be involved in the testing of autonomic functions.
Ethical considerations and regulatory approvalThe trial will be conducted in accordance with the requirements of the latest version of the Helsinki declaration [22] and regulation (EU) 2017/745 of the European Parliament and of the council of 5 April 2017 on medical devices (MDR) [23]. The research objective dictates use of children in the trial and thus, MDR, Article 65 is activated. Therefore, different sets of trial information will be given; a version adapted to the child’s age, a complete version to the guardians and to the patients ≥ 15 years of age. The information will be communicated in an individual meeting by a trial investigator that has experience with ADHD and children. Potential participants will be given at least 24 h to consider if they want to participate. Patients and guardians will be informed that they may withdraw their consent at any time. We will respect the child’s explicit wishes of not wanting to participate regardless of their age. Children will be informed that any concerns or questions they may have regarding the treatment, will be heard and taken seriously. As the Monarch eTNS device currently does not have an updated CE mark, an application for the approval has been sent and approved by the Danish Medicines Agency and Regional Ethics Committee for Region Zealand. The trial was registered on clinicaltrials.gov before randomisation of the first participant (ClinicalTrials.gov ID NCT06655610).
ParticipantsAll patients aged 7 to 17 years with a confirmed ADHD diagnosis are potential eligible for inclusion. The final eligibility will be evaluated based on the inclusion and exclusion criteria described below. The patients who have not been assessed by the Wechsler Intelligence Scale for Children (WISC-IV/V test) during the last three years will be tested with the WISC-V by psychologists from the clinics. A new WISC-V assessment will however not be conducted for children who are performing well in school [24].
Inclusion criteria7 to 17 years of age at the time of trial enrolment.
A clinical diagnosis of ADHD according to criteria for ICD-10: F90.0, F90.1, F90.8, F98.8C [25].
The ADHD diagnosis must be verified by the Diagnostic and Statistical Manual for Mental Disorders (DSM-5) [26] using The Schedule for Affective Disorders and Schizophrenia for School-aged Children (K-SADS) [27].
A total score above 24 on the ADHD rating scale (ADHD-RS) at baseline.
Signed informed consent from parents/legal caretakers and from the patients aged ≥ 15.
We will include treatment-naïve patients, patients who previously have received stimulant medication, and patients in stable, ongoing stimulant medication (methylphenidate or dexamphetamines/lisdexamphetamine) during the time of the trial.
Exclusion criteriaPatients receiving atomoxetine and guanfacine at the time of trial enrolment will be excluded all together due to the impact of these medications on the arousal system, and through this the potential interference with the heart rate variability measurements.
Epilepsy
Electronic or metallic implants.
Serious mental and/or somatic diseases other than ADHD, such as:
Pervasive developmental disorder not including Asperger’s syndrome (ICD-10 F84.0–84.4 + F84.8–84.9)
Schizophrenia/paranoid psychosis (ICD-10 F20-25 + F28-29)
Mania or bipolar disorder (ICD-10 F30 and F31)
Depressive psychotic disorders (ICD-10 F32.3 + F33.3)
Substance dependence syndrome (ICD-10 F1x.2)
Cardio-vascular disorders
Cancer
An IQ below 70 measured by the Wechsler Intelligence Scale for Children [24, 28].
A substantial degree of restless sleep as reported by parents or caregivers and evaluated by the physician.
Other disabilities that may make use of Monarch problematic.
Trial interventionsBoth the active eTNS and sham stimulation will be given by the Monarch eTNS System (NeuroSigma, Inc., Los Angeles CA). Sham stimulation will serve as a control group. Stimulation will be provided by an external pulse generator, placed close to the patient’s bed and attached with thin wires to a disposable patch electrode placed on the forehead. Bilateral stimulation of the first branch of the trigeminal nerve will last for approximately eight hours nightly over the course of four weeks. Patches will be removed every morning. Power will be provided by 9 V lithium batteries, which will be replaced with a newly charged battery every day.
Active eTNSActive eTNS will be provided by applying single, bipolar pulses of 0.5 ms duration at a frequency of 125 Hz, with an active period of 30 s on/off. Stimulation current will range from 0.2 mili ampere (mA) to 10 mA. The level of current, which is noticeable, yet within the level of comfort, will be identified for each patient by titration at baseline. Depending on the perception of stimulation, the level of current may be altered during the four weeks of treatment, by either the guardian or adolescent in control of the settings.
Sham stimulationThe stimulator and patches will be identical in appearance to the active treatment. The guardian/the adolescent will be informed to administer the device in the same fashion as with active treatment. The sham device will however be programmed to only apply stimulation for 30 s every hour during sleep, optimally at a frequency of maximum 2 Hz. As with the active eTNS, the sham stimulation current will range from 0.2 to 10 mA. Such settings have previously been considered to induce the sensation of a current applied to the forehead as seen with active treatment, yet without it being therapeutically effective [29, 30]. Stimulation will be directed through an internal resister, which ensures draining of batteries and need for recharging after each session. The manufacture of the eTNS device (Neurosigma) will oversee the programming the sham device.
Records of interventionPatients and/or their guardians are requested to keep a logbook each night. We will ask them to record the number of stimulation hours and intensity applied, whether stimulation had been interrupted and whether any issues have occurred during that night.
Concomitant treatmentPatients in both groups will have access to one session of psychoeducation on ADHD provided by a psychologist. All families with an interest in ADHD will have access to an educational video on ADHD. Patients are allowed to continue any concomitant medications used at baseline (apart from guanfacine and atomoxetine), granted that dosages and type of medication remains stable throughout the trial. The uses of any type of concomitant interventions will be registered by the families in the logbook. Patients initiating any type of medication at the beginning of the trial will be excluded.
Discontinuation or modification of interventionsCompliance is defined as completing the treatment in 70% of the nights during the four weeks. Abruption of treatment is only accepted if it happens less than four nights in a row. One whole night is considered of a minimum duration of five hours of uninterrupted stimulation (see feasibility outcomes below).
Families are advised to withhold from using the device in case of fever. The families are free to withdraw their informed contest at any time without providing an explanation. The trial investigators are authorised to terminate participation in case the participant is diagnosed with one of the predefined exclusion criteria or if the patients encounter intolerable adverse reactions.
OutcomesFeasibility outcomesFeasibility outcomes will be measured at the end of the 4-week treatment period.
1)The proportion of participants assessed for eligibility who consent to inclusion and randomization
We will compare the number of eligible participants to the number of randomized participants. We will accept a difference above 50% (95% CI 40.2% to 59.6%). A larger difference can impose difficulties with recruitment in a future randomized trial.
2)Compliance with the intervention
We will calculate the number of participants fulfilling treatment for both the group receiving sham and active eTNS. Compliance with treatment will be defined as completing the treatment in 70% (95% CI 58.4% to 81.6%) of the nights during the four weeks. Abruption of treatment can only be accepted if it happens within less than four nights in a row.
3)Acceptability of the intervention
The acceptability of the intervention will be assessed using a semi-structured qualitative interview guide with predefined questions:
How did you feel about administering and controlling the device?
How did your child feel about receiving this type of treatment?
How was it for your child to receive home-treatment with the device in addition to other strategies provided by the clinic?
Did the intervention have any influence on the relationship between your child and the parents/siblings/school/friends?
4)Completion of follow up
Completion of follow-up will be defined as completing the assessment of the primary exploratory clinical outcome at the end of the intervention. The number of participants with completed outcomes will be compared to the number of participants in total. If the number of participants completing this assessment is above 90% (95% CI 82.4% to 97.6%), this will be acceptable for a future full-size trial. If the number of participants completing the assessment is below 75%, this will introduce serious problems with the interpretation of the results.
5)Use of concomitant treatment
Any concomitant treatment or changes in medication will be assessed for each participant and subsequently evaluated for the two groups at the end of the trial.
Safety and adverse eventsParents will be able to report directly to a dedicated investigators not involved in data analysis, with any concerns regarding potential adverse events or other safety issues during the trial. Adverse events will be measured by an adverse events rating scale at the end of treatment [29]. Height, weight and vital signs will be assessed at baseline and again following the four weeks of treatment.
Exploratory outcomesExploratory clinical outcomes will be assessed at baseline and at the end of the four-week treatment period. Some specific outcomes will be assessed weekly in addition to pre – and post assessment, as indicated below. In this feasibility trial, the clinical outcomes will be used firstly in an exploratory manner.
Primary exploratory outcomeADHD core symptoms—measured by the ADHD-IV rating scale (parent- and teacher rated) [31]. Parent ratings will be assessed at baseline, weekly, and at end of treatment. Teacher ratings will be assessed at baseline and at end of treatment.
Absence from school – measured as total hours of absence reported by logbook (parents) and through direct contact with the teachers.
Secondary exploratory outcomesEmotional liability – measured by Conners 3 Global index, Emotional liability subscale (parent – and teacher rated) [32]
Quality adjusted life years—measured by the Child Health Utility instrument (CHU9D) (parent/self-rated) [33]
Functional impairment – measured by the Weiss Functional Impairment Rating Scale (WFIRS) [34]
Overall severity and improvement in symptoms – measured by the Clinical Global Impressions Scale (CGI) [35]
Behavioural and emotional difficulties – measured by the Strengths and Difficulties Questionnaire (SDQ) (parent – and teacher rated) [36]
Cognitive functioning—measured by the Behavior Ratings of Individual Executive Functions (BRIEF) (parent rated). Assessed at baseline. [37]
Sleep quality—measured by The Children Sleep Habits Questionnaire (CSHQ) (parent – and teacher rated) [38]. Assessed at baseline, weekly and at end of treatment.
Exploring the physiological impactAutonomic functions will be assessed using heart rate variability (HRV) analysis. A chest belt capable of measuring heart rate and inter-beat interval (IBI) will be used at baseline and after the end of treatment. Measurements and calculations of analysis parameters will be done according to relevant standards and guidelines [39, 40]. Several HRV parameters can be indicative of a change in autonomous status. Since all of these are based on measured IBI, we will explore a range of parameters and compare results to existing data where such data exist.
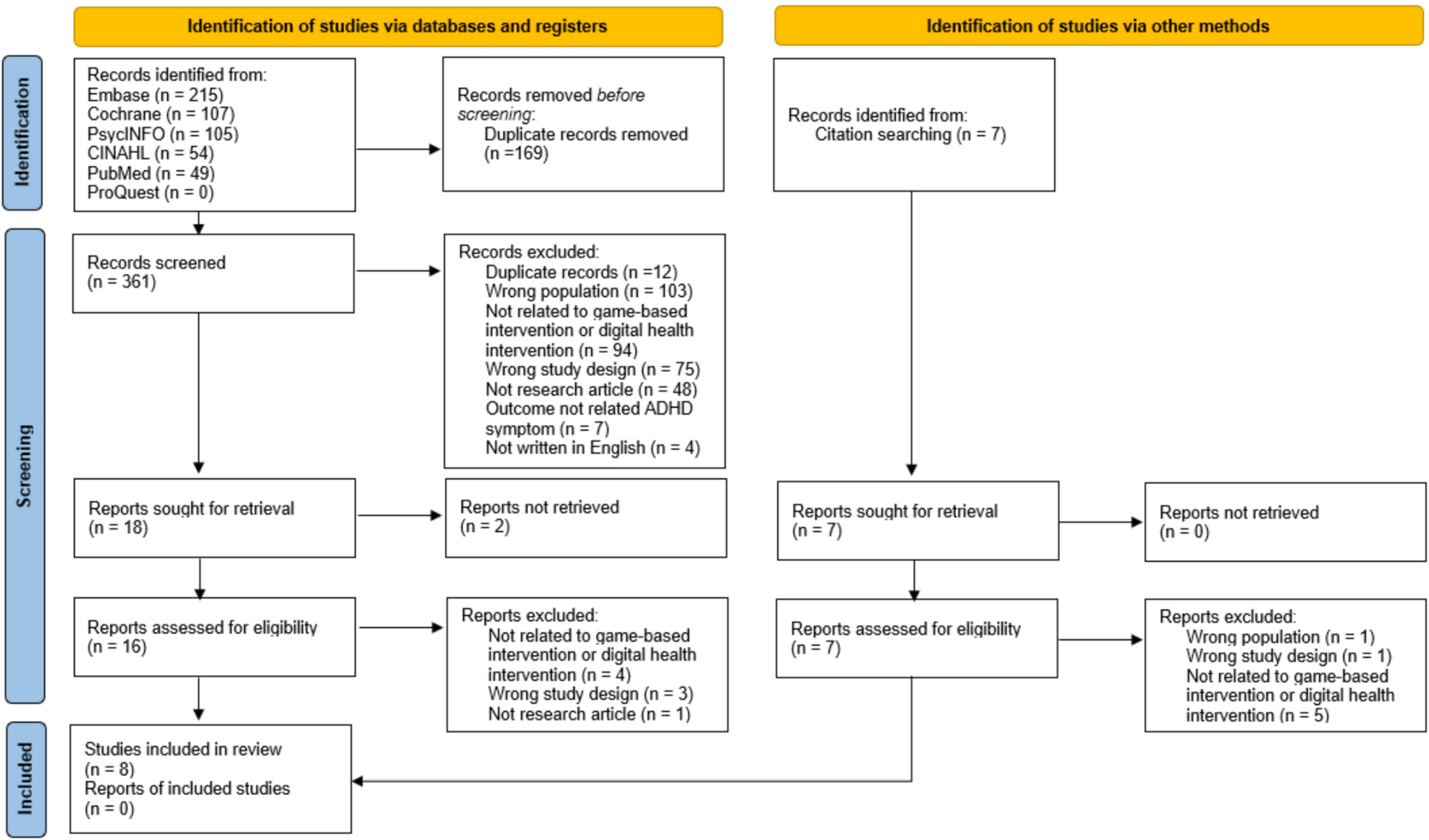
Participant timelineEligible participants will be identified by clinicians working at the respective trial sites. They will ask the families whether they are willing to be contacted by the trial researchers for further information. The trial researcher will send a letter of information and request permission to follow-up with a phone call. Following the phone call, an in-person meeting will be scheduled, in which both oral and further written information will be provided. If the families wish to participate, a second in-person meeting is scheduled. At this second meeting, informed consent is obtained as well as a standard diagnostic—and health evaluation alongside baseline assessment and HRV measurements. The child is finally included in the trial, granted they meet the inclusion criteria following the health – and diagnostic evaluation. Randomisation then commence, in which the participant is allocated to either active or sham eTNS.
They will then proceed to test the device for four weeks. Families will keep a logbook for daily reporting on how the previous night had gone by. Once a week, they will fill in an online questionnaire received by email related to the primary exploratory outcome and the reporting of any adverse events. This email will also contain questions regarding the use of the device and concomitant medication. In addition, the families will receive a weekly phone call from the trial researcher, to remind them to fill in the questionnaire and to allow the families to ask questions if needed. Families may also contact a trial researcher directly via phone throughout the trial period.
Following the four weeks, an in-person meeting is scheduled, in which the families and trial investigators meet to discuss and share their experience. During this meeting, exploratory outcomes will once again be assessed, alongside HRV measurements and the rating of any adverse events. The full participant timeline is seen in Fig. 1.
Fig. 1 Sample size and power consideration
Sample size and power considerationWe will include 60 participants which corresponds to approximately 20% of the sample size needed for the full-scale randomized superiority trial. This corresponds to a power of 48% for the primary outcome (ADHD core symptoms) in this feasibility trial, indicating that the risk of type 1 error is high, and any finding thus may only be considered as exploratory.
Randomisation and blindingThe randomization procedure will be computer-based and provided by The Copenhagen Trial Unit (CTU). The allocation sequence generation will be performed using block sizes of varying lengths concealed for the investigators. Randomization will be stratified by use of ADHD medication compared to no use of ADHD medication at entry. The allocation ratio will be 1:1. Both participants, personal and outcome assessors will be blinded to treatment allocation. Only one investigator will have access to group assignments. This investigator will also be the main person involved with any technical difficulties the families may experience during the trial. Premature disclosure of blinding may occur in case of adverse events where unblinding in considered essential for the safety and further treatment of the patient.
Data managementData will be collected using electronic case report forms (eCRFs) and handled using REDCap (Research Electronic Data Capture) [41, 42].
Data analysisBaseline characteristics will be reported and described using descriptive statistics. Continuous outcomes will be analysed by linear regression and dichotomous outcomes will be analysed using logistic regression. In the primary analysis we will include the intention-to-treat population, i.e. all randomised participants will be included in the analysis in the group to which they were randomised, even when they have received only part of the intervention. The analyses will be adjusted for the stratification variables used in the randomisation. Before the randomisation of the last participant, we will develop and publish a detailed statistical analysis plan with a detailed description of the analyses.
Statistical analyses will be performed by two blinded statisticians presenting independent reports. Our patient population includes both treatment-naïve patients as well as those already exposed to medication. In this feasibility and pilot trial, we will assess the collective effect across all patients included. We will not investigate the effect of intervention based on medication status. Such subgroup analysis will, however, be conducted in the later, larger randomised trial. A data monitoring committee will also be included in the future, larger trial.
Quality assurance and quality controlThe trial will be conducted in compliance with the protocol across all sites. Detailed instructions and Standard Operating Procedures are developed for specific tasks, as needed. The trial will be monitored by internal monitoring. Personnel from the clinical sites will be responsible for monitoring each other’s site respectively.
Comments (0)