Risk minimisation programs are drug safety interventions mandated by regulators to ensure that a product’s benefit–risk profile is positive. Known as ‘additional risk minimisation measures’ in the European Union (EU), and ‘risk evaluation and mitigation strategies’ (REMS) in the USA, these drug safety programs are required to be assessed periodically to determine whether they have been effective in minimising the targeted drug-related risk(s) [1, 2].
1.1 Regulatory Research Standards
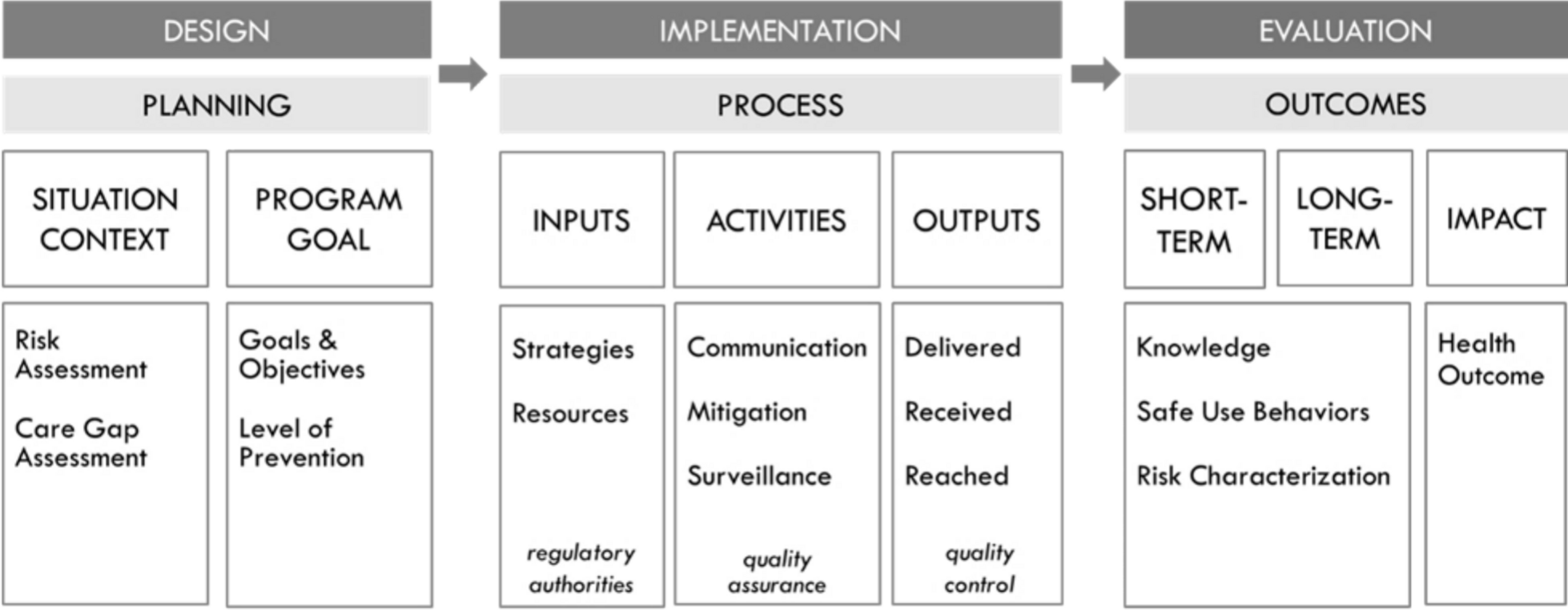
Regulatory guidance regarding risk minimisation program design and acceptable evidence of risk minimisation program effectiveness has been evolving over the past several decades. In the EU, new pharmacovigilance regulations engendered a series of ‘good pharmacovigilance practice’ (GVP) guidelines, replacing previous guidance as of 2012 [3]. In particular, GVP Module XVI, whose third revision was finalised in 2024, describes risk minimisation principles and measures (‘tools’) to use as well as methods for developing and evaluating them [1]. In the USA, the passage of the Food and Drug Administration (FDA) Amendments Act of 2007 stimulated updated guidance on drug safety, including introduction of REMS [4]. In 2019, in response to commitments outlined in the fifth authorisation of the Prescription Drug User Fee Act, the FDA released a draft guidance entitled ‘REMS Assessments: Reporting and Planning’ (referred to hereafter as the REMS Assessment Guidance), which provided information on how to conduct a REMS evaluation [2]. In 2024, a draft guidance for industry was released entitled ‘REMS Logic Model: a Framework to Link Program Design with Assessment’ [5]. The latter guidance set forth a structured, transparent approach to designing, implementing and evaluating risk minimisation programs, one that involves depicting how program context, inputs and activities relate to program outputs and outcomes [5]. The EU GVP outlines an essentially similar approach with an implementation pathway of risk minimisation measures within the medicine’s benefit–risk management cycle.
A comparison of these US and EU guidance documents shows how regulatory perspectives on risk management evaluation have recently converged in terms of six key principles (Table 1):
1.
Use formative evaluation approaches to determine whether the risk minimisation program can be implemented [1, 2].
2.
Consider the context in which the program is to be, or was, implemented when embarking on plans to evaluate it [1, 2, 5].
3.
Seek input on program design, implementation and evaluation from the intended end-users of the risk minimisation program (e.g. healthcare professionals and administrators, patients and their informal caregivers) [1, 2].
4.
Use implementation science theories, models and frameworks (TMFs) to help guide the design, implementation and/or evaluation of a risk minimisation program [1, 2, 5].
5.
Evaluate multiple domains, including the implementation process and its outcomes (including both intended and unintended program effects), using implementation science TMFs to provide guidance on specific domains to evaluate and how to do so [1, 2, 5].
6.
Conduct summative evaluations at multiple timepoints [1, 2].
Table 1 Principles for the design and evaluation of risk minimisation programs congruent with both the European Medicines Agency and United States Food and Drug Administration in risk minimisation guidanceCollectively, these converging principles set forth robust standards for risk minimisation program evaluation. These new standards can be met via the use of mixed methods, that is, qualitative and quantitative data collection and integration—in conjunction with an implementation science TMF. Such an approach can improve the comprehensiveness, depth and consistency of data collected, thus generating more fit-for-purpose information for regulatory decision-making. For example, a mixed methods evaluation could feature quantitative surveys of program end users (e.g. healthcare professionals, patients) to identify contextual factors that are impeding program implementation; qualitative interviews could then be conducted to understand these contextual factors in more depth and to explore possible strategies for addressing them within specific clinical care settings. Survey and interview guide items would be guided by the particular TMF chosen for the study. Both the EMA and FDA cite the value of using mixed methods approaches, guided by implementation science TMFs, for formative as well as summative evaluations [1, 2, 5]. Despite this, few examples of such an approach have been reported in the literature to date [6,7,8,9].
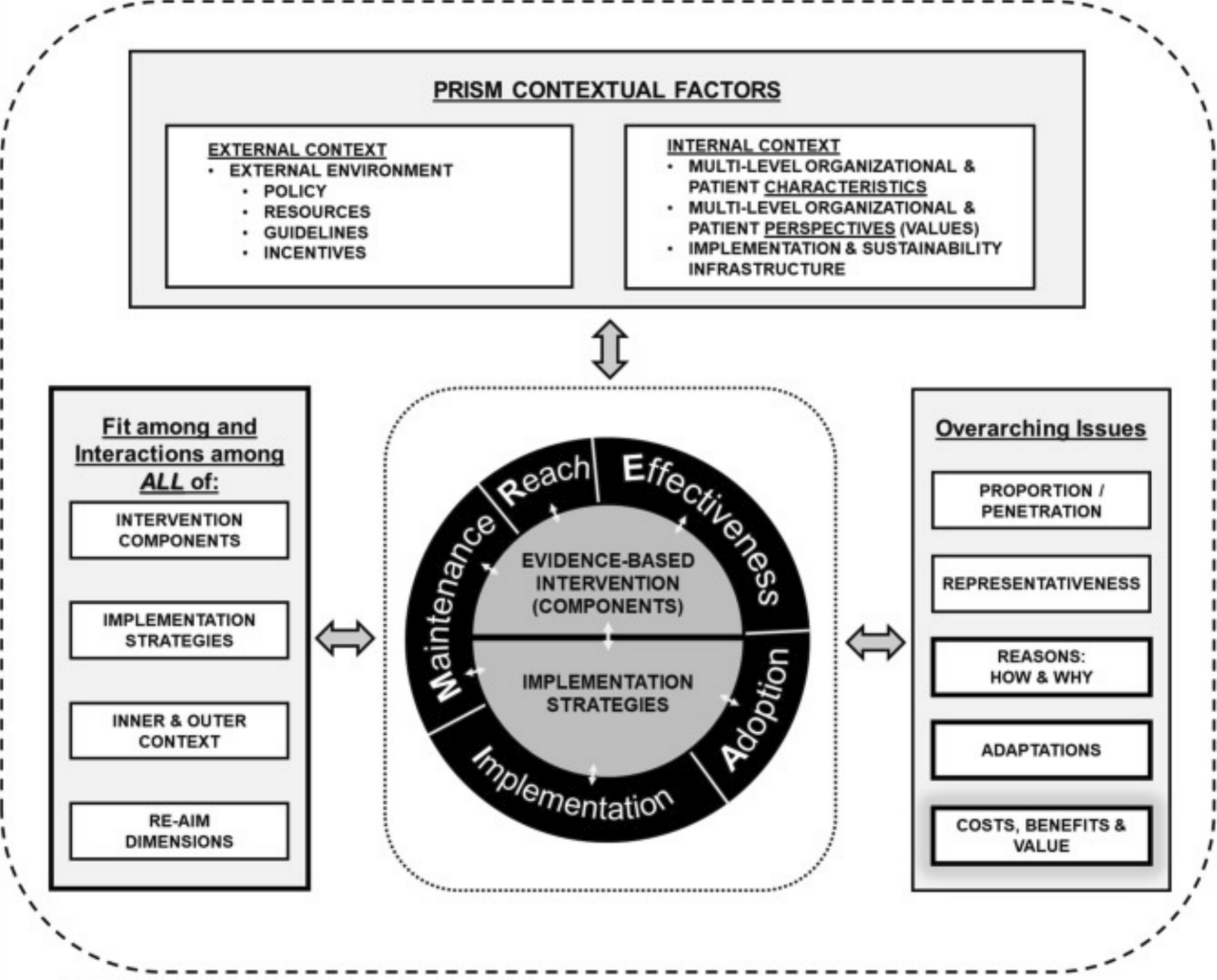
There are multiple types of implementation science TMFs and multiple ways to apply them in risk minimisation evaluation studies [10,11,12,13]. For example, they can be used to specify the mechanism(s) whereby risk minimisation activities are expected to achieve uptake and impact, to plan program implementation and evaluation, to inform the selection of relevant domains and constructs to measure and to guide data analysis. The FDA’s REMS Logic Model provides a systematic, structured approach to incorporate the REMS assessment planning into the design of a REMS [12]. Multiple different implementation science TMFs have been developed for different purposes, and guidance on how to select one to use for a given research project is available [14, 15]. Examples of implementation science evaluation frameworks include the Consolidated Framework for Implementation Research (CFIR) [16], the RE-AIM [17], the Practical, Robust, Implementation and Sustainability Model (PRISM) [18] and the Precede-Proceed Model [19]. Elements of different TMFs can be combined to meet the requirements of a particular research project if needed [10, 20]. In a mixed methods approach, a framework can be used with both quantitative and qualitative methods to obtain complementary information on contextual factors affecting program implementation and effectiveness and to link quantitative ratings and qualitative findings to outcomes.
The EMA and FDA guidances also acknowledge the importance of assessing contextual factors (barriers and enablers) to program implementation and effectiveness as part of the evaluation [1, 2]. Risk minimisation programs are required to be implemented as a condition of marketing authorisation within the responsible regulatory agency’s jurisdiction. Thus, the context or circumstances under which implementation is to occur may vary widely for any given risk minimisation program. A risk minimisation program approved in the EU, for example, may need to be implemented across all 30 countries in the European Economic Area, each differing in terms of its healthcare system, clinical practice standards, language, culture and other contextual factors.
A recent example in the EU, the valproate risk minimisation program, illustrates how context can affect both program implementation and outcomes. Valproate is an effective treatment approved for epilepsy within the EU; however, it also has high teratogenic potential, and infants exposed to valproate in utero are at high risk of birth defects and neurodevelopmental disorders [21]. In light of this, the EU introduced risk minimisation measures for the product in 2014. These measures specified that valproate must not be offered to females of childbearing potential (FOCBP) unless alternative therapies were shown to be ineffective or not tolerated, in which case such patients were required to use contraception and other pregnancy prevention steps were to be imposed [22]. FOCBPs currently on valproate were to be gradually switched to other treatments under appropriate medical supervision [23,24,25]. Despite these measures, however, the program’s effectiveness in reducing the number of exposed pregnancies proved to be modest [23]. At a public hearing in 2017, testimony from healthcare professionals and patients indicated that program implementation had varied markedly by country and healthcare setting, and that in contexts where implementation had been poor, awareness of the risk minimisation measures had been low and the recommended safe use behaviours for both providers and patients had not been consistently adopted [26]. An in-depth analysis of the feedback from patients and healthcare professionals at the public hearing and a further stakeholder event revealed challenges in implementing and disseminating the risk minimisation measures, lack of consensus among healthcare professionals in terms of the therapeutic place of valproate, and the types of risk minimisation measures and training needed, and the degree to which those measures could be integrated into existing healthcare processes [27]. To date, there is no REMS requirement for valproate in the USA.
Additionally, the EMA and FDA guidances recommend that a range of different data sources be employed, including ‘complementary data sources that provide a combination of qualitative and quantitative information’ such as in a mixed methods design [2]. Mixed methods evaluations can mitigate the presence of ambiguities or incompleteness resulting from reliance on quantitative survey data alone, thereby enhancing both the quality and comprehensiveness of the evidence on program feasibility, implementation and effectiveness [28,29,30]. Collecting both quantitative and qualitative data can provide deeper insights into the phenomenon of interest, that is, ‘a whole greater than the sum of the parts’ [31]. Doing so, however, entails an a priori specification of the rationale for mixing the data, the types of data to be collected and analysed, the relative priority to be given to quantitative and qualitative research in the study, the methods sequence (i.e. concurrent or sequential) and the research phase(s) in which the integration or mixing of each data type is to occur (e.g. during sampling, analysis) [28,29,30].
Lastly, EMA and FDA have converged in terms of the specific domains to assess in mixed methods studies evaluating the effectiveness of risk minimisation programs (Table 3). These include the extent to which the program was implemented as designed and reached the intended audience(s) (program ‘coverage’), the level of knowledge of and attitudes about the risk minimisation program among the involved stakeholders, the degree of behavioural change by targeted participants (i.e. extent to which healthcare professionals and patients adhere to the recommended safe use practices set forth in the risk minimisation program), the impact on patient health outcomes and extent to which the program had unintended consequences (e.g. creating barriers to accessing the drug) and posed undue burden on the healthcare system.
1.2 Shortcomings in Risk Minimisation Effectiveness Research
Over the past decade, several systematic reviews have documented significant shortcomings in both the robustness of the methods used to assess these programs (including their overreliance on survey methodology) and in the validity of the evaluation findings [5, 20, 32,33,34,35,36,37,38,39,40]. Specific limitations cited include lack of timely and comprehensive data on implementation outcomes, inadequate evaluation of relevant outcomes, absence of information regarding program adaptations to improve fit within a given context and uninterpretable or inconclusive findings [32,33,34,35,36,37,38,39].
Such inadequacies may be attributable in large part to shortcomings in the risk minimisation evaluation designs that have been recommended and used to date. On the basis of a review of risk minimisation assessment studies registered in the Heads of Medicines Agencies (HMA)/European Medicines Agency (EMA) Catalogues of Real-World Data Sources and Studies [41] over the past decade, we found that the single most frequent type of evaluation (75.4%) has been cross-sectional surveys of healthcare professionals and/or patients (Table 2). Risk minimisation programs can be complex, featuring multi-level and multi-component interventions which require evaluation at multiple levels (e.g. patient, individual healthcare professional, healthcare setting, healthcare system, region or country). While surveys can assess the level of impact of risk minimisation measures at the individual user level (e.g. on safe use knowledge and self-reported behaviour), they fail to provide in-depth information as to why an impact occurred (e.g. factors at the organisational, system and/or country level that may have served to prevent or enable impact), under what circumstances (i.e. contextual factors or conditions) it did so, strategies used to support implementation and the mechanisms linking program context, process and outcomes [33, 34, 36, 42]. It has been repeatedly questioned whether program effectiveness assessments, as designed and conducted to date, are fit-for-purpose in informing regulatory decision-making [5, 20, 32,33,34,35,36,37,38,39,40].
Table 2 Number of risk minimisation evaluation studies listed in the HMA-EMA catalogues of real-world data sources and studies, 2014–2024a1.3 Study Objective
The purpose of this paper was to exemplify how a mixed methods approach, guided by a framework, can be used to design formative and summative evaluations of a risk minimisation program.


Comments (0)