
Remember me




Wilms tumour (WT), or nephroblastoma, is the most frequent renal tumour in children and was first described in 1899 by the German doctor Max Wilms [8].
WT is an embryonal malignancy resulting from disrupted kidney development [9]. During embryogenesis, the intermediate mesoderm forms the metanephric mesenchyme, which transforms into epithelium to create the kidney cells [10]. In the WT, this process is disrupted at various levels, generating mixtures of blastemal, epithelial, and stromal cells, sometimes with myogenic differentiation [11]. The histology of the tumour reflects both genetic defects and times of divergence from normal renal formation [10].
The cause of WT is not completely clear but is thought to be linked to genetic changes in the normal development of the genitourinary tract [8].
Initially, only mutations of WT1, CTNNB1, WTX and loss of H19-IGF2 imprinting were known, explaining only some cases. Other genes associated with WT include TP53 and MYNC [12]. Furthermore, loss of heterozygosity in chromosomes 1p, 1q, 11p15 and 16q may indicate aggressive clinical behaviour [13] but does not clarify the mechanisms of tumor development nor the possible therapeutic targets. Recent analyses have uncovered additional genetic factors, including chromatin modifiers and microRNA genes such as DROSHA, DGCR8, DICER1 and other, involved in renal development [14]. The mutation of these genes is important for therapeutic implications.
WT is the most frequent type of renal tumor in children, but its incidence varies greatly between different regions and ethnic groups [10].
Specifically, in East Asia the annual incidence is lower with 4.3 cases per million children, compared to North America and Europe where 8–9 cases per million are registered [15]. In the United States, African American children have the highest incidence with 9.7 cases per million, while those in the Asia-Pacific Islands have the lowest incidence with 3.7 cases per million [15]. However, estimating the global incidence is difficult due to the lack of childhood cancer registries in some regions and the poor quality of available data, as not all cancers are reported or registered [16]. Furthermore, in areas with fewer resources, around 50% of patients are diagnosed when the cancer has already metastasized [17].
WT is one of the rare childhood cancers that is most frequently found in girls, with a prevalence approximately 10% higher than in boys. In boys, the incidence of this cancer reaches its peak at 1 year of age, with 17.9 cases per million person-years. In girls, however, the incidence remains nearly constant between 1 and 3 years of age, with rates of 17.8, 18.0 and 18.1 cases per million person-years respectively [10].
WT usually manifests as a single lesion, but in approximately 7% of cases it may be multifocal, and in 5–9% of cases it is bilateral [15]. Generally, tumors affecting only one kidney develop at a slightly older age than those affecting both kidneys.
17% of WTs present as part of a well-defined malformation syndrome, and among these, approximately 10% are linked to a specific genetic predisposition for this type of tumor [10].
The genetic syndromes to which WT is associated are WAGR syndrome with a 50% chance of developing the tumor, Denys-Drash syndrome with a 90% chance, Beckwith-Wiedemann syndrome with a 5–10 chance %, and other less common syndromes such as Sotos syndrome, Perlman syndrome, trisomy 18, Frasier syndrome, Bloom syndrome, Li-Fraumeni syndrome and Simpson-Golabi-Behmel syndrome [8].
Clinically, in most children, WT manifests as a palpable asymptomatic abdominal mass that is first noticed by chance by a family member or primary care physician. Other symptoms present in 20–25% are hypertension, hematuria, and flank pain, and presentation after blunting abdominal trauma with abdominal/side pain and blood loss has also been observed [18].
Subsequently, to confirm the diagnosis of WT it is essential to proceed with imaging tests and laboratory tests.
An abdominal ultrasound (US) is often the first test used to identify a kidney mass. If necessary, a magnetic resonance imaging (MRI) or computed tomography (CT) scan may also be done to get more precise images and to assess whether the tumor has spread to the lymph nodes or other organs. A chest X-ray (XR) may be done to check for metastases in the lungs, as this is the first site of metastases.
Laboratory tests evaluate renal function, electrolyte levels and complete blood count, urinalysis, coagulation studies, and cytogenetic studies for the search for 1p and 16q deletion.
In some cases, a renal biopsy is required to confirm the diagnosis of WT, to administer appropriate pre-operative chemotherapy, and some tissue is stored frozen for molecular studies. However, the diagnosis is often made on the basis of clinical and radiological characteristics, and the tumor is surgically removed.
Staging of the tumor is critical to understanding how far the cancer has spread and to planning treatment. The National Wilms Tumor Study Group (NWTSG) staging system is commonly used, classifying the tumor from Stage I, which is limited to the kidney and completely removed, to Stage V, which is present in both kidneys.
PathologySamplingThe nephrectomy specimen is photographed, measured, and inked. After opening, samples of tumor and normal kidney tissue are taken for biological studies. At least one longitudinal slice of the cancer is sampled, with additional blocks from different areas. In the case of a multicentric tumor, each nodule is sampled. The interface between tumor and normal kidney, renal and tumor capsule, and renal sinus involvement are examined for staging. The remaining kidney, hilar fat, and all lymph nodes are also sampled for metastases.
MacroscopyMacroscopically, WT usually presents as large masses altering renal contours, varying significantly in size.
Upon cutting, the surface is heterogeneous in many cases, with areas of viable tumor, hemorrhage, and necrosis (Fig. 1), especially in pretreated specimens. The viable tumor is usually light gray to slightly pink solid in consistency or yellow grey in soft consistency. Some tumors are cystic and careful investigation for solid foci is necessary.
To avoid contamination, it is important to sample the hilar margins, including vessels, if possible before cutting the tumor. Multicentric tumors are present in 5% and are usually associated with nephrogenic rests [19, 20].
Fig. 1
Wilms tumor (A and B) On section, the surface is heterogeneous, with areas of viable tumor, hemorrhage, and necrosis
HistologyWT classically presents a triphasic histological pattern, composed of epithelial, stromal and blastemal components, and the proportions and degree of differentiation can vary, giving rise to a large variety of tumor presentations. Biphasic and monophasic variants are less frequent.
Preoperative chemotherapy, according to the protocol of the International Society of Pediatric Oncology (SIOP), can considerably modify the original histology of the tumor, reducing or increasing some elements or inducing their maturation.
The blastema is the least differentiated and most malignant component of WT, characterized by small, round blue cells with overlapping nuclei and high mitotic activity. There are different histological models of blastema: diffuse, serpentine, nodular and basaloid. The serpentine pattern presents broad bands of undifferentiated cells surrounded by fibromyxoid stroma, whereas in the basaloid variant the blastema cords have a peripheral palisade of elongated cells with epithelial differentiation.
All these patterns can coexist in the same tumor and have no prognostic value, but their recognition is useful to differentiate the tumor from other “small round blue cell tumors” when only the blastemal component is present.
Blastemal-type WT, often with diffuse growth, may present marked infiltration without a pseudocapsule.
Primitive tubular epithelial structures in blastemal nodules may appear to resemble neuroblastoma with pseudorosettes. Additionally, the appearance of epithelioid or spindle cells may vary depending on early differentiation. There are no definitive criteria to distinguish blastema from early epithelial differentiation or the stromal lineage, and classification of WT subtypes is based on subjective morphological criteria.
The epithelial component of WT displays various stages of differentiation, ranging from primitive epithelial rosette-like structures to tubules and glomeruli-like structures, reflecting different stages of nephrogenesis. Examples of heterologous differentiation include squamous epithelial islands and mucinous epithelia.
The stromal component may include both densely packed undifferentiated mesenchymal cells and looser myxoid cell areas, which may or may not be difficult to distinguish from chemotherapy-modified nontumorous stroma. In some tumors, especially after preoperative chemotherapy, a heterologous differentiation of the neoplastic stroma, which includes smooth or skeletal muscle cells, adipose tissue, cartilage, bone and even glial tissue, can be observed.
Chemotherapy-induced modification may include necrosis, hemorrhage, fibrosis, and hemosiderin-laden macrophages. The blastemal component, highly proliferative, responds well to chemotherapy, while the mature epithelial and stromal components are less sensitive and show a reduced response to preoperative therapy.
It is important to highlight that classification and assessment of response to chemotherapy vary between SIOP and NWTS/COG, influencing risk stratification and treatment. Fully necrotic tumors are considered low risk, while tumors with more than 10% blastema are classified as mixed [19, 21].
Furthermore, the SIOP uses the terms “stromal type” or “epithelial type” to classify pretreated tumors, whereas the NWTS/COG describes them as “stromal-predominant” or “epithelial-predominant.”
Anaplastic WT (AWT) constitutes 5–7% of cases of this type of renal tumor [22]. The diagnosis of anaplasia is based on the presence of large, atypical mitotic figures, and enlarged, hyperchromatic nuclei.
These tumors are typically aneuploid, and the anaplasia may be focused or diffuse. Focal anaplasia indicates a limited area with anaplastic characteristics, while diffuse anaplasia is considered an unfavourable histological feature and can negatively influence the prognosis [22].
Subsequently, SIOP introduced a new risk stratification, and focal AWT was classified as intermediate-risk WT, whereas diffuse AWT remained a high-risk tumor [23].
AWT often express the protein p53 and have mutations in the TP53 gene, which are associated with a worse prognosis. MYCN gene dysregulation has also been associated with unfavourable outcomes in cases of anaplastic histology [19].
Nephrogenic rests (NRs) are abnormal areas of embryonic tissue that persist beyond 36 weeks of development and are present in 30–44% of WT kidneys [19]. The term “nephroblastomatosis” was introduced in 1961 and indicates a lesion composed of immature renal tissue.
There are two major types of NR: perilobar (PLNR) and intralobar (ILNR). The PLNR is located at the periphery of the renal lobules, while the ILNR is located in the central part of the lobe and tends to develop earlier.
Nephroblastomatosis is characterized by the presence of multifocal NRs and can present in different histological forms such as incipient, dormant, regressive, sclerotic, obsolescent and hyperplastic.
It is crucial to distinguish NR from WT, as clinical management differs significantly. NR typically does not have a fibrous pseudocapsule, which is almost always present in WT, but this distinction can be complicated in patients treated with preoperative chemotherapy. In some cases, the term “nephroblastic process, consistent with WT or NR” is used until further radiological-pathological data are available.
TreatmentTreatment planning by a multidisciplinary team with experience in treating children with WT consisting of a pediatric surgeon and/or a pediatric urologist, a pediatric radiation oncologist, a pediatric oncologist and a pathologist is essential to define the appropriate treatment.
Most clinical studies on WT in children are conducted by COG RTC and SIOP, with different approaches (Table 2). Specifically, the COG RTC uses immediate surgery for unilateral tumors, while the SIOP begins with preoperative chemotherapy. Both include postoperative chemotherapy and, in advanced stages, risk-based radiotherapy, and infants younger than 6 months are treated with primary nephrectomy [24].
The COG RTC group, which includes the former NWTS group, has established the standard of treatment for WT in North America. This approach involves an initial nephrectomy, followed by chemotherapy and, in some cases, radiation. This method allows for a timely and precise histological diagnosis, the collection of biological materials not influenced by the therapy and the evaluation of staging, such as the presence of tumor leakage or lymph node involvement, before chemotherapy [25,26,27].
SIOP uses preoperative chemotherapy for patients with renal tumors before definitive resection. This method reduces tumor leakage during surgery and lowers the postoperative stage. Comparing histological features, preoperative chemotherapy alters tumor histology, reducing blastemal and mixed histology types, and results in fewer stage III tumors than immediate surgery [25,26,27].
Table 2 Wilms tumor treatmentStage ITreatment options for stage I WT (Table 2) patients vary based on tumor histology and patient-specific characteristics [24].
Children older than 24 months with tumor weighing more than 550 g treatment involves nephrectomy with lymph node sampling, followed by the chemotherapy regimen with vincristine and dactinomycin × 18 weeks after nephrectomy [28].
Patients with loss of heterozygosity on 1p/16q treatment involves nephrectomy with lymph node sampling, followed by the chemotherapy regimen with vincristine, dactinomycin and doxorubicin × 24 weeks [28].
Patients with focal anaplastic tumor treatment involves a nephrectomy with lymph node sampling, followed by a chemotherapy regimen with vincristine, dactinomycin and doxorubicin × 24 weeks and radiotherapy [28].
Patients with diffuse anaplastic cancer, treatment involves a nephrectomy with lymph node sampling, followed by a chemotherapy regimen with vincristine, dactinomycin and doxorubicin × 24 weeks and radiotherapy [28].
Stage IITreatment of stage II WT (Table 2) involves several standardized options depending on the histology of the tumor [24].
For patients with favourable histology, the standard treatment consists of a nephrectomy accompanied by lymph node sampling, followed by the chemotherapy regimen with vincristine and dactinomycin × 18 weeks after nephrectomy [28].
In the case of patients with loss of heterozygosity on chromosomes 1p and 16q, the chemotherapy regimen with vincristine, dactinomycin and doxorubicin × 24 weeks [28].
For tumors with focal anaplastic histology, treatment involves nephrectomy and lymph node sampling, followed by abdominal radiotherapy and a chemotherapy regimen of vincristine, dactinomycin, and doxorubicin × 24 weeks [28].
Patients with diffuse anaplastic histology receive nephrectomy and lymph node sampling, followed by abdominal radiotherapy and chemotherapy regimen with vincristine, doxorubicin, cyclophosphamide, carboplatin, and etoposide × 30 weeks and with radiotherapy [29].
Stage IIITreatment of stage III WT (Table 2) involves a combination of surgery, chemotherapy and radiotherapy, with different options based on the histological type of the tumor and the presence or absence of particular genetic abnormalities, such as loss of heterozygosity of chromosomes 1p or 16q [24].
In all patients with favourable histology, the standard treatment involves nephrectomy with lymph node sampling, followed by abdominal radiotherapy and chemotherapy regimen with vincristine, dactinomycin, doxorubicin × 24 weeks [28].
Favourable histology with loss of heterozygosity of 1p and 16q, treatment consists of nephrectomy, lymph node sampling, abdominal radiation therapy and chemotherapy regimen with vincristine, dactinomycin, doxorubicin, cyclophosphamide and etoposide with subsequent radiation therapy [30].
In patients with focal anaplastic histology, it comprises two options. Post-operative treatment with lymph node sampling followed by abdominal radiotherapy and chemotherapy regimen with vincristine, dactinomycin and doxorubicin × 24 weeks [28]. Pre-operative treatment with chemotherapy regimen with vincristine, dactinomycin and doxorubicin × 24 weeks [28] followed by nephrectomy and abdominal radiotherapy.
Patients with diffuse anaplastic histology includes two options. Pre-operative treatment with vincristine, doxorubicin, cyclophosphamide, etoposide × 24 weeks [28] followed by nephrectomy, lymph node sampling and abdominal radiotherapy.
Immediate post-operative treatment with lymph node sampling, abdominal radiotherapy, and chemotherapy regimen with vincristine, doxorubicin, cyclophosphamide, carboplatin, and etoposide × 30 weeks + radiotherapy [30].
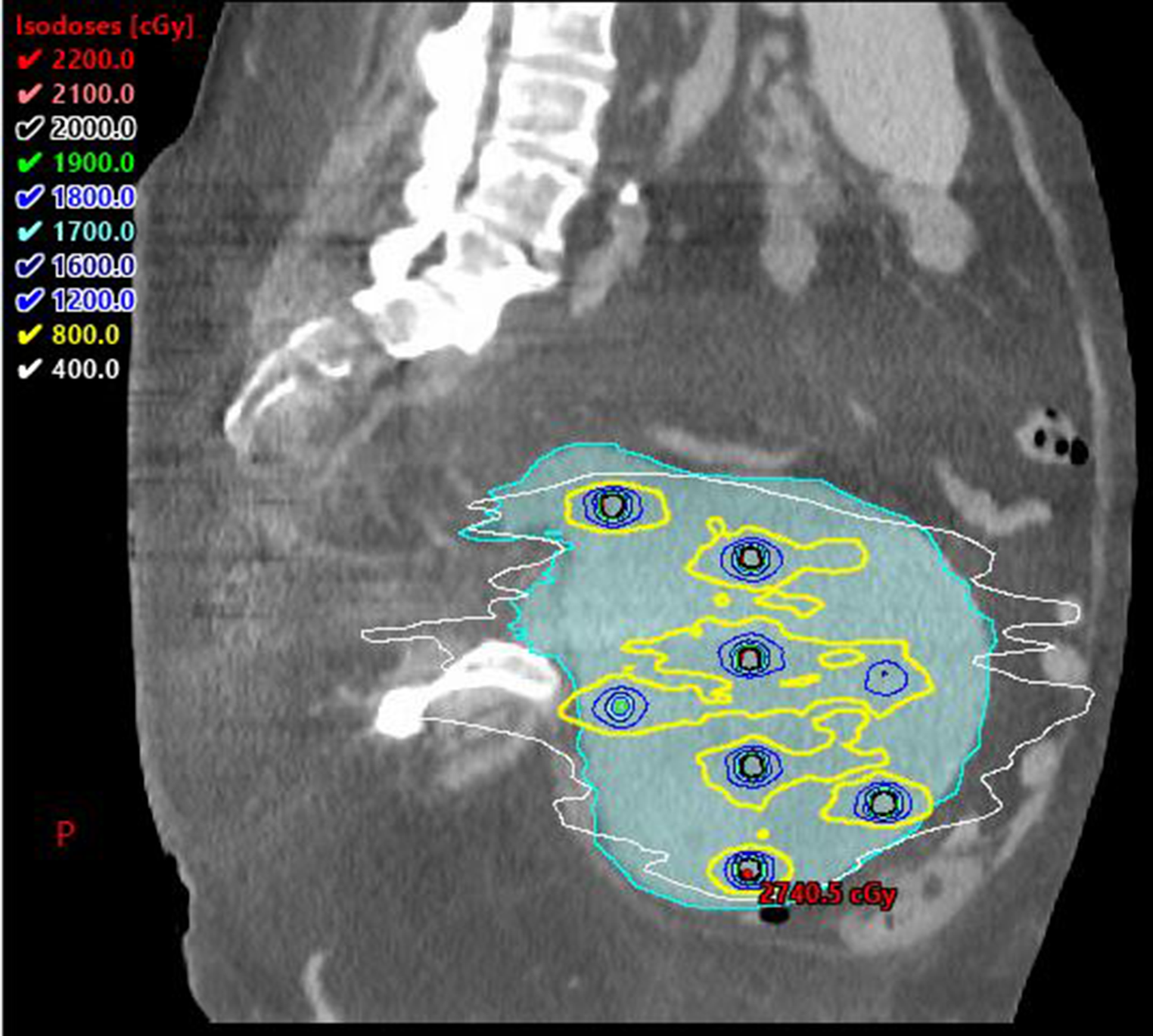
Timely initiation of radiotherapy within 14 days is fundamental for the multimodal treatment of patients with non-metastatic WT, delay is associated with an increased risk of mortality [24].
Negative lymph nodes and the absence of loss of heterozygosity are predictors of excellent survival rates, whereas patients with positive lymph nodes at the time of nephrectomy have a worse prognosis [24].
Stage IVTreatment of stage IV WT (Table 2) varies depending on histological features and the presence of specific genetic alterations such as loss of heterozygosity on 1p/16q or gain of 1q [24].
Options include nephrectomy with lymph node sampling, abdominal and lung radiotherapy, and chemotherapy with regimens such as vincristine, dactinomycin, doxorubicin × 24 weeks [28] or vincristine, dactinomycin, doxorubicin, cyclophosphamide and etoposide with subsequent radiotherapy [30] in case of favourable histology with isolated lung nodules. If focal anaplasia regimen vincristine, dactinomycin, doxorubicin × 24 weeks [28]. If diffuse anaplasia regimen vincristine, doxorubicin, cyclophosphamide, etoposide × 24 weeks after nephrectomy [31].
Pulmonary radiotherapy may be avoided in some patients, depending on the initial response to chemotherapy. Liver metastases are not an independent adverse prognostic factor, and the use of liver surgery remains controversial.
Stage VThere is no standard approach for the treatment of bilateral WT stage V. One study has proposed guidelines, aiming to eliminate tumors and preserve kidney tissue to reduce the risk of chronic kidney disease [32].
Patients with bilateral cancer have worse survival rates than unilateral ones. Treatment options include preoperative chemotherapy, surgical resection, and, in some cases, kidney transplant. Nephron-sparing surgery is preferable to preserve renal function. Postoperative chemotherapy is customized based on tumor response and histology.
New Therapeutic PerspectivesNew therapies for WT (Table 2) include molecularly targeted agents, immunotherapies, and genetic and epigenetic interventions [33, 34].
Molecular Targeted TherapyThe insulin-like growth factor 2 (IGF2) signaling pathway is closely linked to WT development [33]. Overexpression of IGF2, caused by loss of function of the DIS3L2 gene or mutations in some miRNAs that regulate PLAG1, contributes to the formation of this tumor. IGF1R, the IGF2 receptor, is considered a major therapeutic target due to its role in tumor growth. IGF1R inhibitors, such as BMS-754807 and NVP-AEW541, are being tested and show potential in reducing tumor growth in animal models [34].
WT requires angiogenesis to grow and spread, so the VEGF/VEGFR pathway is a common therapeutic target, with drugs such as apatinib, bevacizumab, and AZD2171 being used or tested to reduce the density of tumor blood vessels [34]. Control of WT1 could modulate the efficacy of therapy. However, anti-angiogenic therapy can cause serious side effects, such as pneumothorax, particularly in children [34].
The PI3K/AKT pathway is important for cell proliferation and survival, and abnormal activation may favor WT [33]. Inhibitors like buparlisib and everolimus can inhibit tumor growth, although buparlisib has high toxicity [34]. Modulating regulators such as PTEN and KRAS could offer new therapeutic strategies.
ImmunotherapyImmunotherapy aims to control and eliminate tumor cells by restoring or enhancing the normal immune response against the tumor.
Inhibition of COX-2 could be crucial in the treatment of WT. In mouse models, COX-2 is highly expressed in the tumor microenvironment, resulting in infiltration of immunosuppressive cells and production of cytokines such as IL-10 and TGF-β [35]. These factors promote tumor immune escape and thus COX-2 inhibition could be crucial in the treatment of WT.
CAR-T cell therapy uses CRISPR/Cas9 gene editing to modify T cells, allowing them to recognize and attack specific cancer cells. There are several generations of CAR-Ts, each with improvements in antigen recognition and immune response. However, the application of CAR-T for Wilms tumor is still in the experimental stage and the most promising target for it is B7-H3 [35, 36].
MTAA-CTL is an advanced cell therapy that uses CD8 + NK-T cells and CTLs to fight tumors more effectively than CAR-T therapy. This therapy uses dendritic cells modified with tumor-specific antigens to target multiple tumor antigens at the same time. Clinical trials have shown that it is safe and can improve response in WT patients, although further studies are needed to confirm its specific efficacy for this tumor [35, 37].
Genetic and Epigenetic TherapymiRNA is a non-coding RNA that regulates gene expression post-transcriptionally by binding to mRNA and influencing translation or degradation of mRNA. Studies have shown significant differences in miRNA expression profiles between Wilms tumor cells and normal kidney tissues [34].
Several miRNAs such as miR-21, miR-19b, miR-483-3p, miR-891b, miR-613, miR-140-5p and miR-572 have been associated with the onset and development of WT [38, 39], and some of these miRNAs promote tumor growth, while others have suppressive effects [40].
Comments (0)