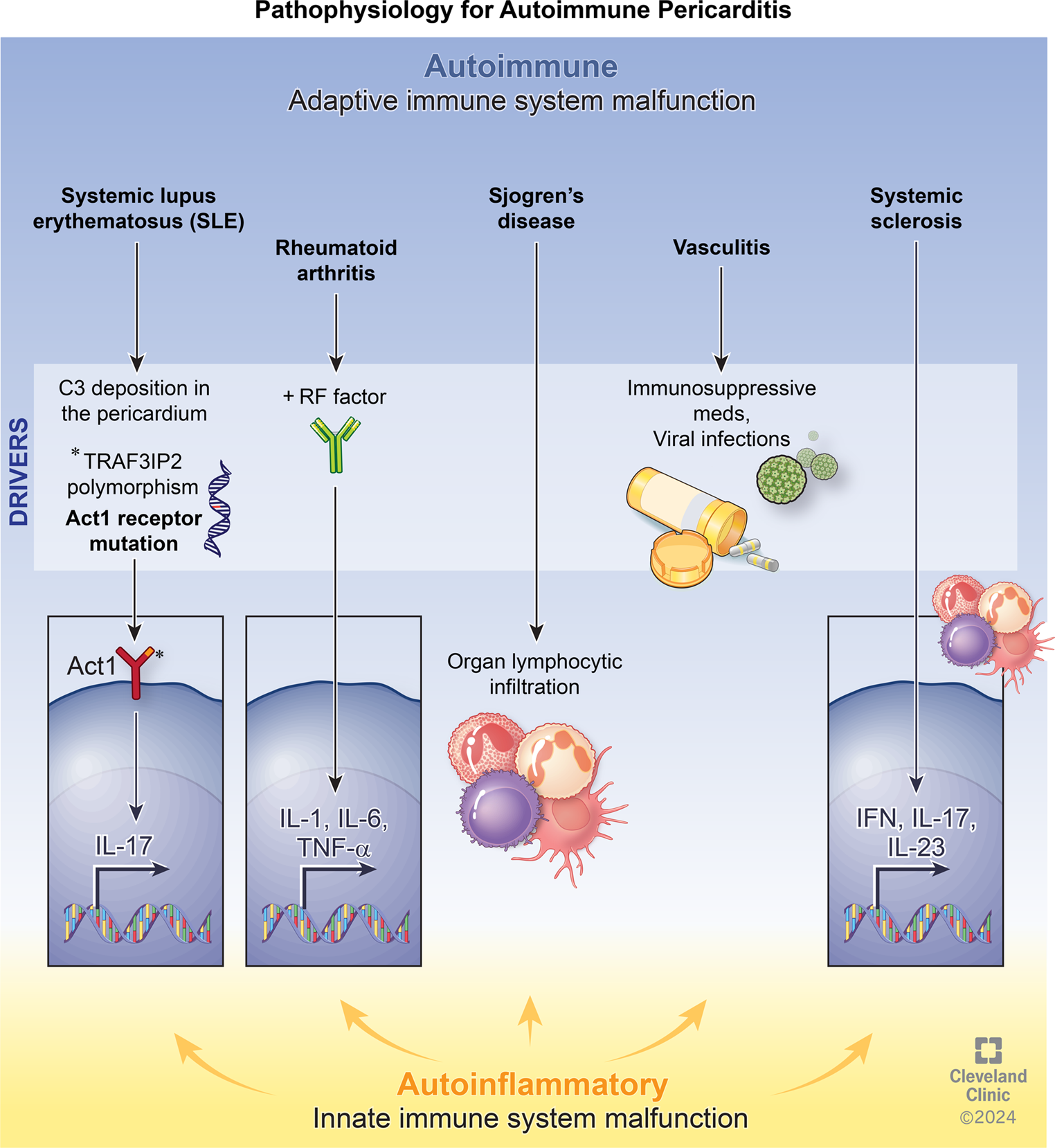
Dealing with new biomarkers and their diagnostic and predictive value, we need to explore the pathogenesis of IRAP. Different pre-clinical and clinical studies discovered a potential interplay between innate and adaptive immunity in the pathophysiology of pericarditis, describing a relationship between autoimmune and autoinflammatory mechanisms. Autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, Sjogren’s syndrome, Behçet’s disease, inflammatory bowel diseases and vasculitic processes may be complicated by pericardial effusion and pericarditis [13,14,15]. The positive response to glucocorticoids or immunosuppressants represents another important clue supporting the hypothesis of an adaptive immunity imbalance in relapsing pericarditis [16]. Different infectious (cardiotropic viruses, bacteria and other microbial hosts) and non-infectious (surgery, ischemia, irradiation, trauma, bleeding) triggers [17], interplaying with other genetic and environmental factors [18], could induce and contribute to direct or indirect damage in pericardium layers: viral infections can trigger an autoimmune response against the pericardium due to antigen mimicry. In addition, non-infectious injuries can lead to an immune response by exposing or releasing cardiac autoantigens. These processes activate B and T lymphocytes, mainly Th1 and Th17, leading to the release of interleukin (IL)-6, IL-8, and interferon (INF)-γ and possible production of anti-nuclear (ANA), anti-heart (AHA) and anti-intercalated disk (AIDA) autoantibodies, which stimulate proinflammatory damage [17, 19]. Cytokines are detectable only in pericardial fluid as a local pro-inflammatory reaction, whereas autoantibodies are checked in serum and pericardial fluid [20, 21]. A possible differential diagnosis could be made using cytokine signatures in pericardial fluid, where high tumour necrosis factor (TNF)α and low transforming growth factor (TGF) β1 levels correlate with viral pericarditis, whereas a low IL-6 concentration with autoreactive pericarditis [21]. An interesting prognostic aspect of adaptive immunity is a reduction of naive-T cells and the overexpression of activated CD8 + T effector correlate with a more active refractory inflammatory response; on the other hand, a higher number of regulatory T cells and normal expression of naive T and activated CD8 + T cells should suggest a controlled inflammatory phenotype [22]. Further studies could be useful to assess the reliability of these markers in diagnosis and prognosis.
More recently, a correlation between IRAP and autoinflammatory diseases has been speculated: a dysregulation of the inflammasome, a large intracellular multiprotein complex in neutrophils and macrophages, might overproduce pro-inflammatory cytokines such as IL-1 and TNFα. Recurrent pericarditis occurs in the setting of different genetic autoinflammatory diseases, particularly in tumour necrosis factor receptor-associated periodic syndrome (TRAPS), familial Mediterranean fever (FMF) and, rarely, mevalonate kinase deficiency, in which mutations in genes involved in the inflammatory response provide an overexpression of several pro-inflammatory cytokines [23]. The best-characterized inflammasome has a sensor molecule called NLR pyrin domain-containing 3 (NLRP3), activated in several inflammatory conditions, including gout, atherosclerosis and pericarditis [24,25,26]. The inflammasome is activated by binding of pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) with specific membrane or intracellular receptors, such as Toll-like receptors (TLRs) and NOD-3 receptors, and high IL-1 concentration is released into the site of injury, with recruitment of important effector cells, mainly neutrophils, and enhancement of inflammation [27, 28]. In this context, the early stages of IRAP show a predominantly neutrophilic microenvironment, along with other immune cells and exfoliated and disintegrated mesothelial cells [16]. Neutrophils have a pivotal role in autoinflammatory diseases and IRAP [29, 30]. They can destroy cell debris and microbes through a process called phagocytosis but also release many pro-inflammatory mediators such as IL-1, IL-1 receptor antagonist, IL-6, IL-12, TGFβ, TNFα, oncostatin M and B lymphocyte stimulator (BLYSS) to activate themselves and other immune cells, mainly macrophages [29, 31]. Additionally, neutrophils create neutrophil extracellular traps (NETs), web-like structures made of cytosolic and granule proteins assembled on a scaffold of decondensed chromatin, which neutralize pathogens but, if dysregulated, they can induce the production of IL-6 and pro-IL-1β [32]. Moreover, neutrophils interact with platelets by releasing chemokines such as CC-chemokine ligand 5 (CCL-5) and platelet factor 4 (PF4); these molecules promote further neutrophil and monocyte adhesion, NETosis, and the secretion of granules, developing microthrombosis in a complex mechanism known as “thromboinflammation” [29, 31].
Table 1 summarizes the principal biomarkers in pericarditis, their utility and limitations.
Table 1 Summary of principal biomarkers for diagnosis and prognosis in pericarditis: utility and limitations*C-reactive Protein (CRP)
CRP is the primary and most important acute-phase protein found in the serum [33]. Its levels rise as a non-specific response to infectious and non-infectious inflammatory processes. This protein is produced only by hepatocytes and is mainly controlled by cytokines, particularly IL-6, which is released in the bloodstream during inflammation. Its serum concentration is determined only by its synthesis rate, according to the severity of the inflammatory stimulus. Once the stimulus ceases completely, the circulating CRP concentration drops rapidly, almost at the same rate as the plasma CRP clearance [34]. In a prospective study, high-sensitivity CRP (hs-CRP) was found elevated in 78% of 200 patients at the onset and during recurrences of IRAP. Among patients with normal CRP levels at 24 h from the onset of symptoms (22% of 200), 34% of them showed an increase when tested later and another 50% of them had either received anti-inflammatory treatment previously (that might impair CRP increase, particularly corticosteroids and anti-IL1 agents) or not re-evaluated afterwards. In another retrospective study on 202 patients, hs-CRP levels were elevated in 76% of patients within 6 h after onset; the percentage rose to 96.7% when retested after 24 h and 98.7% after 48 h [35]. In fact, CRP has its kinetics (as troponins, for instance): after a single stimulus, CRP concentrations in the serum increase rapidly to more than 5 mg/L in about 6 h, reaching a peak around 48 h; the plasma half-life of CRP is approximately 19 h and remains constant under all conditions of health and disease [34]. Interestingly, in acute myocarditis, CRP is often stably normal or near normal and we might speculate that CRP is lower in cases of associated myocarditis [36,37,38]. If the initial result is negative in suspected pericarditis, we recommend to retest for CRP after 12–24 h. Overall, 15–20% of cases of acute pericarditis have persistently normal CRP [39]. The pathogenesis of these forms with low-grade inflammation is not known. CRP is driven mainly by IL-6. However, in viral infections and autoimmune diseases characterized by the Type I interferon gene signature, CRP seems to be an unreliable marker of inflammation: in fact, the levels of CRP in the bloodstream can be relatively low, despite the presence of significant inflammation, as indicated by elevated IL-6 levels. Moreover, different CRP individual responses may be genetically regulated [40].
Pericarditis with elevated CRP usually shows clear signs of inflammation, such as fever, pericardial and pleural effusion, high white blood cell counts with neutrophilia and lymphopenia and elevated ESR; to date, pleuropulmonary involvement is highly predictive of the inflammatory phenotype [35, 39]. These patients are treated more frequently with pericardiocentesis and anti-IL1 drugs.
CRP also has a predictive value. High CRP levels in IRAP at onset are associated with major in-hospital cardiac complications, such as cardiac tamponade, non-obstructive cardiogenic shock, ventricular tachycardia, large symptomatic pericardial effusion requiring pericardiocentesis or pericardiectomy, up to death [35].
Either CRP or hs-CRP can be tested. We did not notice relevant differences between the two tests when used in patients with pericarditis. This is likely because hs-CRP is mainly used to reliably detect minimal elevations (e.g. 0.2 vs. 0.4 mg/dL), primarily associated with cardiovascular risk [41]. On the other hand, non-hs-CRP tests are useful in acute inflammatory conditions, where more pronounced elevations are observed (e.g. values higher than 1 mg/dL, often 10–20 mg/dL). In other words, for values in the high range, such as 1 mg/dl or higher, both test are reliable and reproducible. After treatment begins, it is important to monitor CRP levels to assess the effectiveness and duration of treatment with non-steroidal anti-inflammatory drugs (NSAIDs), colchicine, steroids, and anti-IL1 agents [6, 7, 42,43,44]. Typically, CRP levels normalize slowly (weeks) after treatment with NSAIDs [36], but quickly, in a few days, with corticosteroids and anti-IL1 drugs, usually anakinra [45] or rilonacept [44]. In most cases (85%), hs-CRP normalizes within 2 weeks of treatment. However, hs-CRP remains elevated in 40% of patients after the first week and in 15% after the second week, necessitating prolonged therapy of more than 2 weeks [36]. During the initial acute phase, we recommend regularly monitoring CRP levels on a weekly basis to track disease activity. It is important to avoid reducing the initial drugs doses too early, as this could lead to a reactivation of pericarditis while CRP levels are still high. We suggest considering tapering off the medication only when CRP levels have completely normalized.
Persistent elevation of CRP longer than 1–2 months in our opinion should be considered as a risk factor for a specific aetiology, not yet identified, or for an incessant course and recurrence, along with other factors such as fever > 38 °C, corticosteroid use and incomplete response to medical therapy [46, 47]. To gradually taper treatment, it is crucial to regularly monitor both the signs and symptoms of the disease, as well as inflammatory markers, especially CRP levels, typically on a monthly basis. Specifically, lowering the prednisone dosage should only be considered if the patient is symptom-free and their C-reactive protein level are normal [7].
Complete Blood Cell Counts (CBC) with Differential Formula
In cases of IRAP, like in autoinflammatory diseases, white blood cells rise, with significant increases in absolute and relative numbers of neutrophils and decreases in absolute and relative numbers of lymphocytes, leading to a higher NLR [10, 39, 48]. The NLRP3-produced IL-1β induces the proliferation of granulocyte precursors (promyelocytes, myelocytes, metamyelocytes) in the bone marrow and the recruitment of neutrophils into the peripheral blood [10, 39] and pericardium [49], suggesting a direct involvement of neutrophils in the serosal tissue [29, 30]. In a study by Kim et al. in pericardial fluid, patients who developed effusive-constrictive pericarditis after pericardiocentesis showed elevated concentrations of leukocytes with a higher percentage of neutrophils and a lower percentage of monocytes [49]. Indeed, during both the first attack and recurrences, these haematological alterations correlate with a marked systemic acute-phase response, as evidenced by CRP and ESR levels, along with pleural and possible peritoneal effusion [10]. Increased CRP and neutrophil leukocytosis should not be mistakenly considered signs of bacterial infection in this context, prompting an escalation of antibiotic therapy [10, 39, 50]. To this regard, we have recently provided data from a large cohort of patients that demonstrated that pericarditis with a systemic inflammatory phenotype is often a manifestation of a systemic autoinflammatory disease, most frequently with a pleuropulmonary involvement and that this specific pericarditis phenotype shows similarities with other autoinflammatory diseases [39]. To date, IRAP shares clinical and laboratory similarities with the inflammatory syndromes where, beyond periodic fever, increased CRP and polyserositis, high NLR have been demonstrated [48, 51, 52].
Counts of specific types of white blood cells, particularly neutrophils and lymphocytes, as well as the neutrophil-to-lymphocyte ratio (NLR), can serve as valuable prognostic markers for various medical conditions, including stroke [53, 54], acute myocardial infarction (AMI) [55, 56], sepsis [57].
Peripheral blood differential counts have a predictive value in IRAP: relative neutrophilia, absolute and relative lymphopenia with high NLR can indicate a higher risk of developing 12-month recurrences [10]; the NLR and CRP are also independent predictors of 12-month recurrences and cardiac tamponade [58]. Lastly, as CRP levels, complete blood cell count with differential formula may be used to monitor the response of treatment.
Immature granulocytes, which measure the number of promyelocytes, myelocytes, and metamyelocytes in peripheral blood, can serve as a novel biomarker for increased bone marrow activation and inflammation [59]. It could be an indicator of severity and mortality in different acute conditions such as sepsis [60], acute pancreatitis [61], upper gastrointestinal bleeding [62] and coronary artery diseases (CAD) [63]. Selvi et al. found a high percentage of immature granulocytes in patients with acute pericarditis. A value higher than 65% has high sensitivity (80%) and specificity (90.6%) levels [9].
Another blood cell type involved in the inflammatory process, platelets, is not well defined in IRAP. Further research should be implemented to define their role in IRAP. In our clinical experience, hemoglobin levels may decline rapidly during IRAP, with transient normocytic anemia, followed by a rapid rebound at remission, with a median hemoglobin reduction of 1.4 g/dL. Hemoglobin reduction was associated with CRP elevation [64]. In this regard, IRAP may represent a pathogenetic model for this type of anemia, mediated by hepcidin activity [65].
D-Dimer (D-D)
D-dimer (D-D) is a specific product of fibrin degradation. Increased levels of this biomarker may occur in patients with different thrombotic disorders [66]. Despite the low specificity [67] and interference of various physiological and pathological factors [66], D-D testing is currently common in the emergency department to rule out life-threatening conditions such as pulmonary embolism or acute aortic syndrome [68,69,70]. Moreover, D-D can provide insights into other clinical conditions, including disseminated intravascular coagulation, acute coronary syndromes (ACS) and ischemic stroke [71]. D-D elevation was found in a significant proportion (50.6 to 73.3%) of patients diagnosed with acute pericarditis [12,





Comments (0)