Study Design
This study (WAL4001) was an open-label, single-arm, multicenter phase 4 study conducted at five study sites in China. Enrolled patients received ibrutinib orally at a dose of 420 mg once daily until disease progression or unacceptable toxicity. Patients who progressed on treatment were followed up on subsequent anticancer therapy, occurrence of other malignancy, survival status recorded until death, loss of follow-up, consent withdrawal, or study closure. The study protocol was amended once on March 25, 2020, to update the safety information regarding cerebrovascular incidence identified in the post-marketing setting and to clarify procedures and the collection of data.
This study was conducted in accordance with the principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and local applicable regulatory requirements. All patients provided written informed consent according to the local requirements prior to enrollment. The trial protocol and any amendments were reviewed and approved by an independent ethics committee at each investigation site (Supplementary Table S1). This trial is registered with ClinicalTrials.gov (NCT04042376).
Patients
Key eligibility criteria included ≥ 18 years of age with centrally confirmed clinicopathological diagnosis of WM in accordance with the consensus panel of the Second International Workshop on WM (IWWM) [15]. Enrolled patients must also have at least one prior therapy, documented disease progression, or had no response to the most recent treatment regimen, a symptomatic disease meeting at least one criterion from the Second IWWM requiring treatment, measurable disease defined as serum monoclonal IgM > 0.5 g/dL, hematology and biochemical values within protocol-defined limits, and an Eastern Cooperative Oncology Group performance status (ECOG PS) score of ≤ 2.
Patients were excluded from the study if they had known central nervous system involvement by WM, prior exposure to ibrutinib or other BTK inhibitors, hypersensitivity reaction to ibrutinib or to the excipients in its formulation, previously received WM-related therapy at most 30 days prior to first administration of the study treatment, received allogeneic hematopoietic stem cell transplant or plasmapheresis less than 35 days prior to the initiation of study treatment, history of stroke or intracranial hemorrhage within 6 months prior to enrollment, known bleeding disorders or hemophilia, or clinically significant cardiovascular disease, hepatic impairment, or active infection. A full account of the inclusion and exclusion criteria is described in Supplementary Table S2.
Outcomes and Assessments
The primary endpoint was major response rate (MRR, overall response rate was the term stated in the protocol) by investigator assessment, defined as the proportion of patients who achieved partial response (PR) or better according to the modified consensus criteria from the Sixth IWWM [16]. Secondary endpoints were overall response rate (ORR [clinical response rate was the term used in the protocol], defined as minor response [MR] or better), very good PR or better rate, duration of response (DOR), time to response (TTR), PFS, overall survival (OS), pharmacokinetics, and safety. All treatment responses were evaluated as per the Sixth IWWM by investigators [16]. The definitions of the efficacy endpoints are described in Supplementary Table S3.
Safety was evaluated on the basis of adverse events, clinical laboratory tests, physical examinations, and vital signs, summarized using frequencies and percentages. Adverse events were graded per National Cancer Institute Common Terminology Criteria for Adverse Events v4.03 and assessed from informed consent to 30 days after last dose of study drug. Adverse events of special interest defined by the sponsor (major hemorrhage event) were also reported. Patients were observed for any adverse events until 30 days after the last dose of study drug.
Baseline tumor assessment was performed up to 42 days before the first dose of treatment. Computed tomography (CT) scans of the neck, chest, abdomen, and pelvis were performed. Magnetic resonance imaging was allowed if the CT scans could not be used to evaluate the sites of disease adequately. Radiographic imaging was performed every 16 weeks for the first 2 years, and thereafter every 24 weeks until disease progression. Bone marrow aspirate and biopsy were required at screening. Bone marrow was required to confirm complete response (CR), pre-dose at weeks 49 and 97 to assess response, and at time of progression or suspected progression due to progressive cytopenia.
Disease progression based on changes in IgM levels determined either by serum protein electrophoresis or by a quantitative IgM serum immunoglobulin test was confirmed by a subsequent evaluation within at least 4 weeks of the first finding.
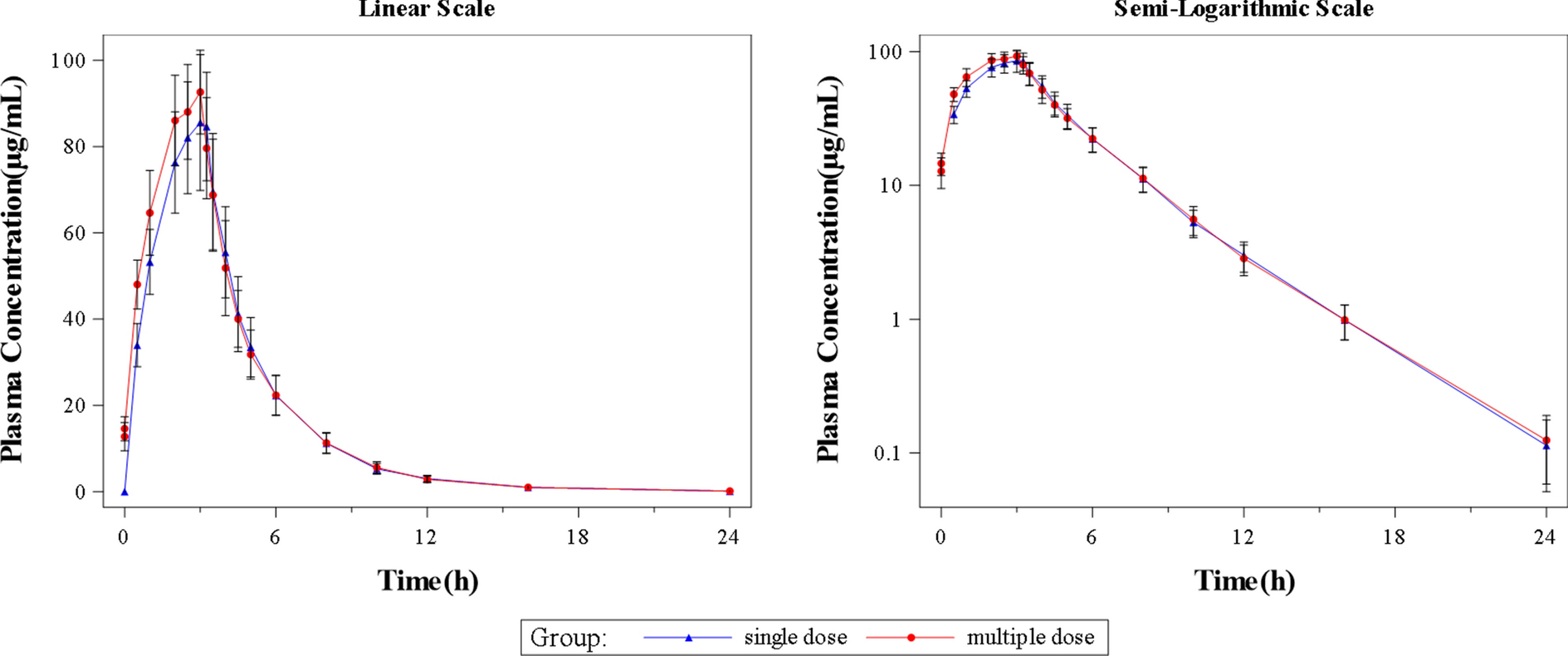
Blood samples were collected from all patients for the measurement of plasma concentrations of ibrutinib and the metabolite PCI-45227 before treatment on day 1 of weeks 1, 5, and 9. The pharmacokinetic-evaluable patients were defined as patients who received at least one dose of study drug and had at least one quantifiable pharmacokinetic sample obtained post-treatment.
Statistical Analysis
Sample size was determined with the assumption that MRR by investigator is 69.8% in the study population, similar to the study by Treon et al. [6]. Hence, approximately 17 patients were recruited to obtain a power of 85% and to achieve a clinically meaningful MRR of 32% or higher with a one-sided significance level of 0.05.
Efficacy and safety analyses were performed on all patients who received at least one dose of ibrutinib. Pharmacokinetics analyses were performed on all patients who received at least one dose of ibrutinib and had at least one available pharmacokinetic sample after treatment. MRR, ORR, and very good PR or better rate were calculated using exact binomial distribution, and the exact (Clopper-Pearson) 90% confidence intervals (CIs) are presented. Time to event variables (DOR, PFS, and OS) and the corresponding 95% CIs were described using the Kaplan–Meier method. Time to major response was summarized descriptively for responders. Observed plasma concentrations of ibrutinib and metabolite PCI-45227 were summarized using descriptive statistics. Exposure matrices, including maximum plasma concentration at steady state (Cmax) and area under the plasma concentration–time curve during 24 h after dosing at steady state (AUCτ,ss), of ibrutinib from this study and from the two pivotal studies (PCYC-1118E and PCYC-1127-CA) were derived using the individually predicted ibrutinib concentrations from popPK modeling.
PopPK Modeling
A popPK analysis was conducted to characterize the PK of ibrutinib in Chinese patients with WM after oral administration. This analysis included data from the current study and the other two global pivotal trials (PCYC-1118E and PCYC-1127-CA) [6, 8]. PK-evaluable patients were defined as those who received at least one dose of study drug and had at least one quantifiable PK sample obtained post-treatment. The concentrations of metabolite PCI-45227 were not included in the popPK analysis.
A popPK model for ibrutinib was first developed using data from patients with B cell malignancies [17]. This initial popPK model was further updated by including additional internal clinical studies in patients with B cell malignancies (updated model, not published) and has been used for this work. In the previous popPK analysis, ibrutinib concentrations could be described using a two-compartmental model with sequential zero to first-order absorption and first-order elimination. The model was parameterized in terms of relative bioavailability and zero to first-order absorption (duration of zero order input [D1], temporal delay [lag time] before absorption process is started [ALAG1], and first-order absorption rate constant [ka]), linear clearance (CL), volume of distribution in the central compartment (V2), intercompartmental clearance (Q), and volume of distribution in the peripheral compartment (V3) (Supplementary Fig. S1). Interindividual variability was modeled with an exponential term. Residual variability was described using an additive model, as the plasma ibrutinib concentrations were log-transformed. The effects of CYP3A inhibitors (taken or not taken) and increasing age (continuous covariate) on F1 and meal condition (fast, modified fast, or fed) on D1 had been identified as statistically significant (but not clinically relevant) covariates for the final popPK model.
In this popPK analysis, the empirical Bayesian estimation (EBE) was performed using the previously developed model on the data from all three studies. In the EBE, basic goodness-of-fit plots were generated to assess the fit of the previous model on the current data, with the following graphs compared: observed concentrations versus population prediction (PRED), observed concentrations versus individual prediction (IPRED), conditional weighted residual (CWRES) versus PRED, and CWRES versus time since first dosing. In addition, a visual predictive check (VPC) was also performed to evaluate the agreement between the observed and predicted concentration generated by the previous popPK model. After we demonstrated that the previous model could describe the current data, the exposure metrics (Cmax and AUCτ,ss) at the recommended dose schedule (420 mg orally once daily) were derived from the predicted ibrutinib concentrations using the individual parameters obtained in the EBE for the three studies. The derived exposure metrics were compared between Chinese patients in the WAL4001 study and non-Chinese patients in the PCYC-1118E and PCYC-1127-CA studies. The analysis was conducted using a nonlinear mixed-effects model analysis in NONMEM® (Version 7.4, Icon Development Solutions, Ellicott City, MD).
In the final dataset, a total of 546 (WAL4001, 31; PCYC-1118E, 62; PCYC-1127-CA, 453) post-dose quantifiable samples of ibrutinib concentrations from 126 (WAL4001, 17; PCYC-1118E, 16; PCYC-1127-CA, 93) PK-evaluable patients were included in the popPK analysis.





Comments (0)