
Remember me
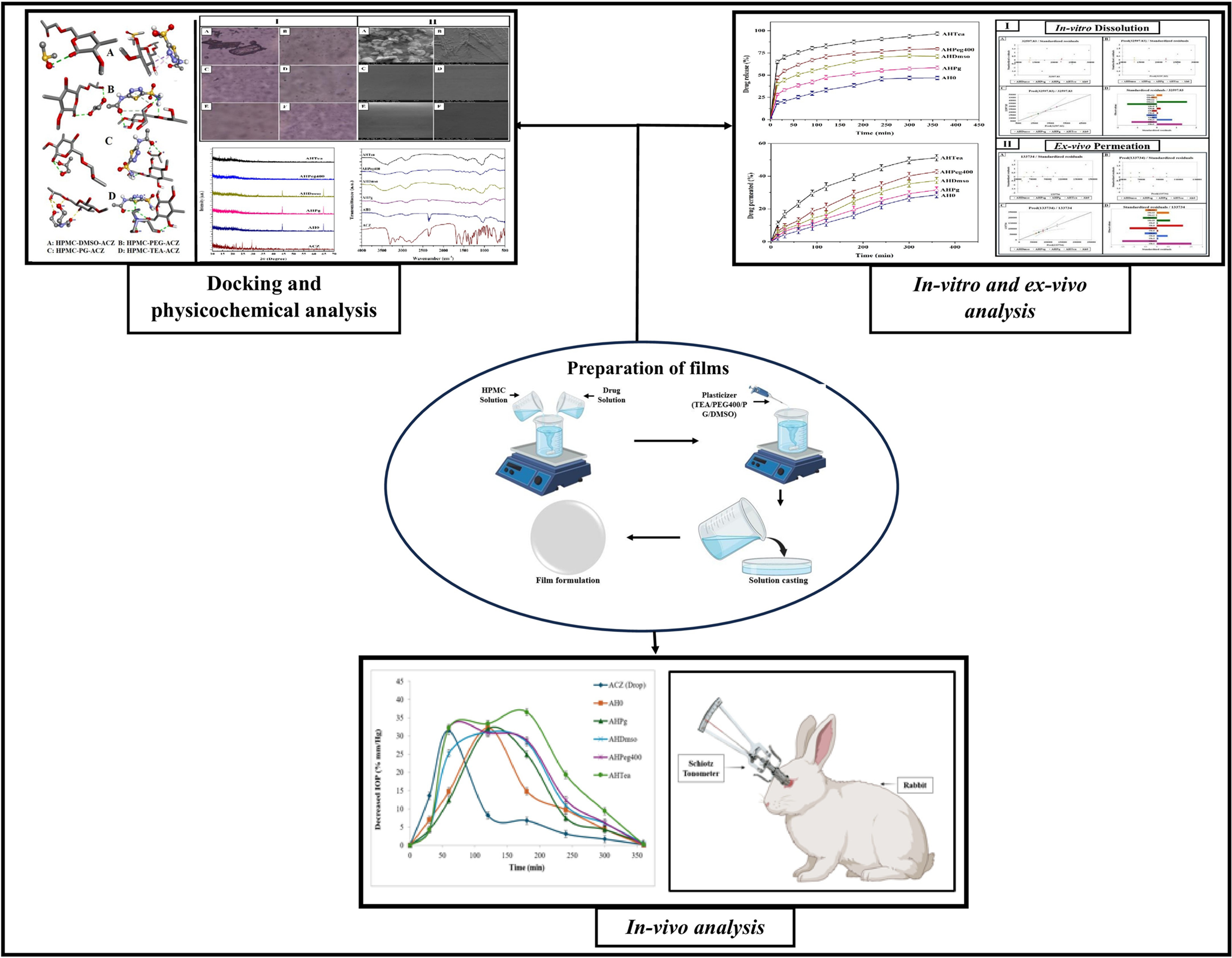
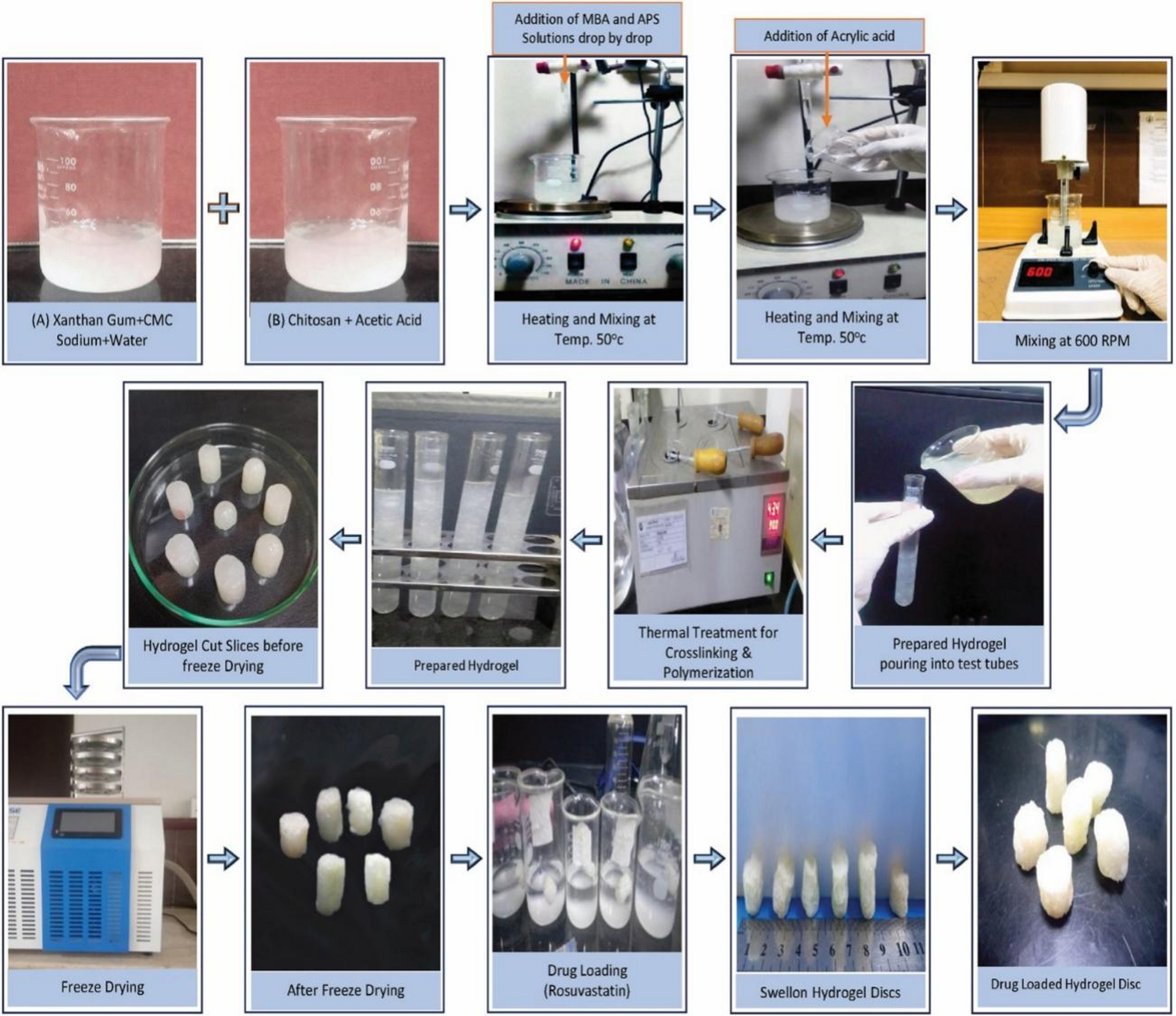
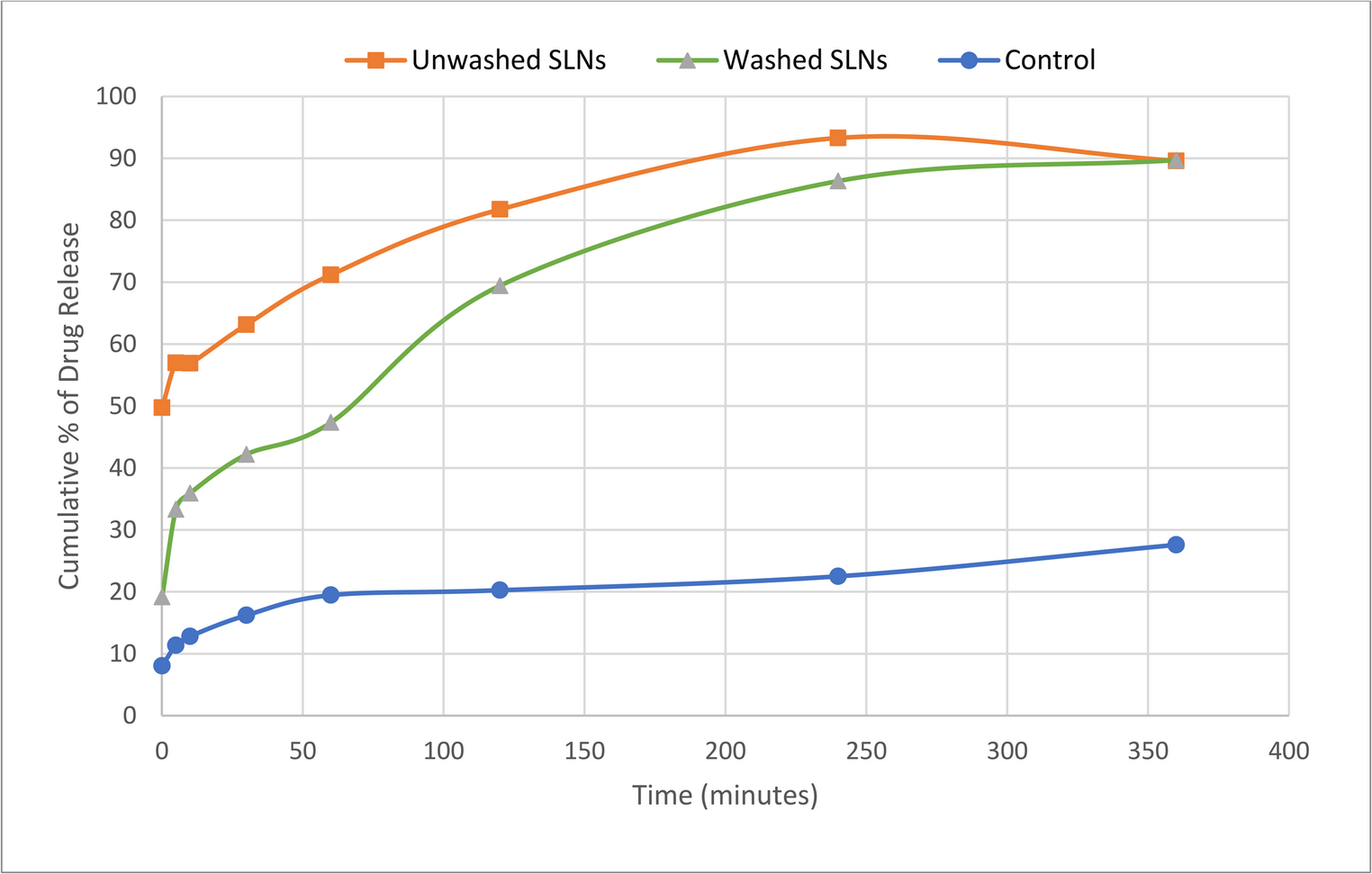
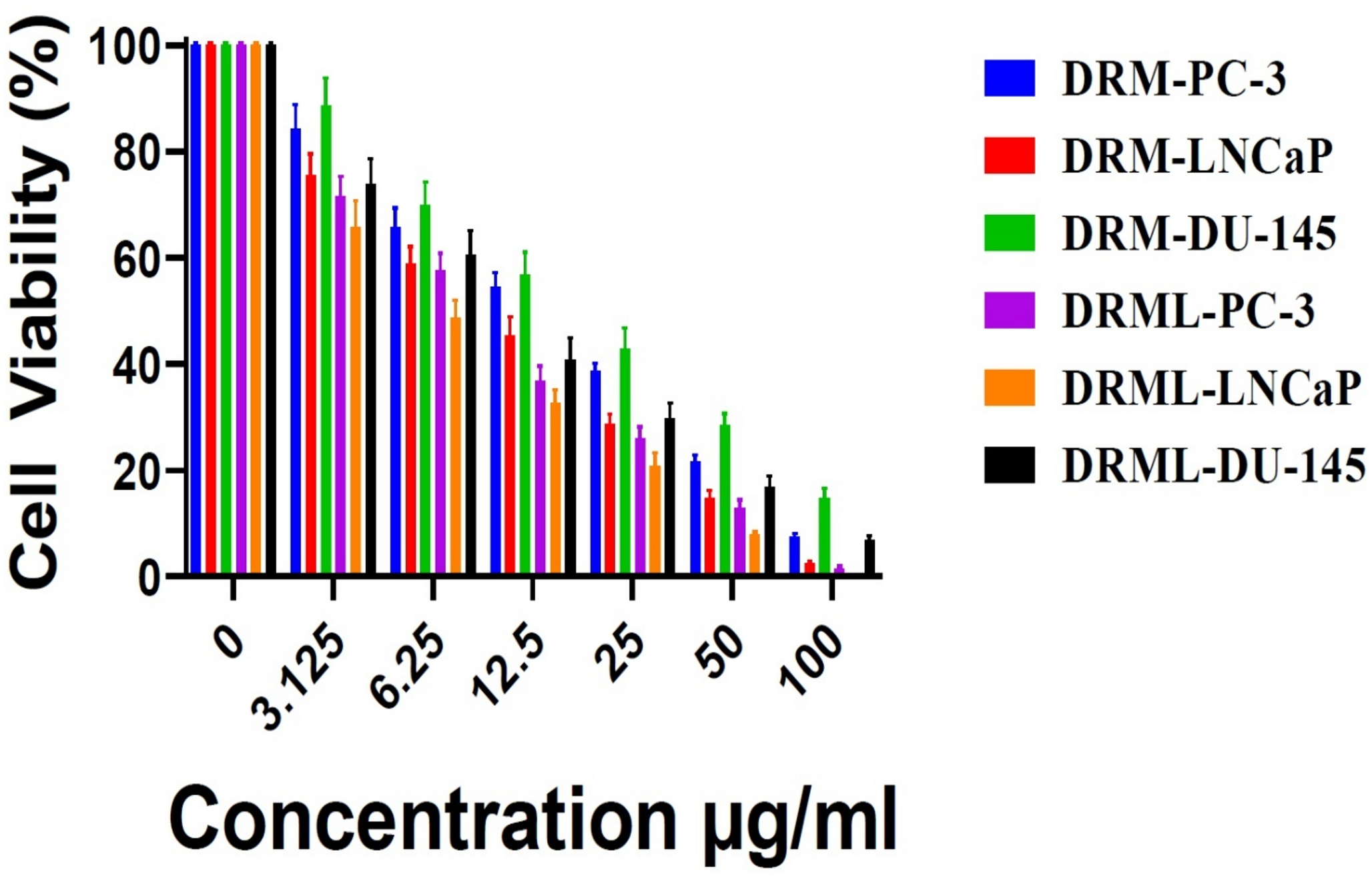

Fungal infections have emerged as a worldwide health burden, affecting millions of individuals globally each year [1]. They range from superficial infections of the skin, nails, and mucosa to invasive systemic diseases associated with high morbidity and mortality [2, 3]. Opportunistic fungi such as Candida, Aspergillus, Cryptococcus, and Mucorales have become increasingly prevalent in immunocompromised populations [4, 5], particularly among patients with HIV/AIDS [6], organ transplant recipients [7], individuals with malignancies [8], and those receiving prolonged immunosuppressive therapy [9]. In addition, the recent rise in antifungal resistance poses a serious therapeutic challenge, with limited drugs available to combat multi-resistant fungal strains [10, 11]. Consequently, the management of fungal infections continues to be a pressing concern in modern medicine.
Historically, the treatment of fungal diseases has been difficult due to the eukaryotic similarity between fungi and human host cells, which restricts selective drug targeting and leads to dose-limiting toxicity [12, 13]. The discovery of amphotericin B in the 1950 s revolutionized antifungal therapy, offering broad-spectrum activity but at the cost of severe nephrotoxicity [14]. The subsequent development of azole antifungals in the 1970 s and 1980 s, such as ketoconazole, fluconazole, and itraconazole (ICZ), marked a new era by targeting ergosterol biosynthesis with improved safety profiles [15, 16]. Later, the introduction of echinocandins in the early 2000 s provided additional therapeutic options, especially for invasive candidiasis, by inhibiting β-(1,3)-D-glucan synthesis in fungal cell walls [17, 18]. Despite these advances, the antifungal pipeline remains relatively limited compared with antibacterial agents, underscoring the urgent need for novel strategies to enhance drug delivery and efficacy [19].
Within the triazole class, posaconazole (PCZ) has emerged as a third-generation broad-spectrum antifungal with superior activity against Candida, Aspergillus, Cryptococcus, and the notoriously resistant Mucorales [20, 21]. Its mechanism of action, potent inhibition of lanosterol 14α-demethylase disrupts ergosterol synthesis, thereby compromising fungal membrane integrity [22]. PCZ is widely used for prophylaxis and treatment of invasive fungal infections, particularly in high-risk patient groups [23]. Despite its strong pharmacodynamic profile, PCZ suffers from poor aqueous solubility, low and variable oral bioavailability, and high lipophilicity, which significantly restrict its therapeutic effectiveness [24]. Moreover, conventional formulations often fail to achieve adequate drug concentrations at localized sites of infection such as the skin, ocular tissues, or vaginal mucosa, highlighting the limitations of current delivery strategies.
The field of nanotechnology-based drug delivery has rapidly advanced over the past two decades, offering innovative solutions to the long-standing challenges of antifungal therapy [25]. Nanocarriers, including liposomes, solid lipid nanoparticles, nanostructured lipid carriers, polymeric nanoparticles, dendrimers, micelles, and nanoemulsion, have been extensively explored to enhance solubility, improve stability, prolong circulation time, and enable targeted delivery of antifungal agents [26,27,28]. Importantly, nanocarriers can facilitate site-specific delivery by penetrating biological barriers such as the stratum corneum, mucosal epithelium, or ocular tissues, thereby achieving high local drug concentrations while minimizing systemic toxicity [29].
For PCZ, the incorporation into advanced nanocarriers represents a particularly promising strategy. Novel formulations designed for topical, systemic, vaginal, and ocular delivery can overcome pharmacokinetic limitations, enhance therapeutic outcomes, and address unmet clinical needs. For instance, topical and transdermal systems enable sustained dermal penetration for cutaneous and subcutaneous fungal infections [30], vaginal formulations improve mucosal retention and patient compliance in recurrent vulvovaginal candidiasis [31], while ocular nanocarriers enhance corneal permeation to effectively treat fungal keratitis [32]. Such approaches not only improve patient-centered care but also represent a paradigm shift in antifungal drug delivery.
Scope and Rationale of the ReviewAlthough several reviews have discussed antifungal nanocarriers or PCZ formulations in a general context, most provide descriptive summaries that are either limited to a single delivery route or focus primarily on physicochemical optimization without considering route-specific biological barriers and translational feasibility. Moreover, existing reviews often emphasize systemic delivery, while localized administration strategies remain fragmented across the literature.
The present review addresses this gap by offering a strategic, route-oriented evaluation of advanced nanocarrier systems incorporating PCZ for topical, ocular, vaginal, and systemic delivery. It systematically compares how nanocarrier composition, architecture, and preparation methods are tailored to overcome tissue-specific challenges, enhance localized antifungal efficacy, prolong residence time, and minimize systemic exposure and toxicity. In addition, this review uniquely links critical formulation attributes (such as particle size, entrapment efficiency, surface charge, and release behavior) with biological and therapeutic outcomes, providing mechanistic insight beyond conventional descriptive reporting.
Furthermore, the review extends beyond formulation performance to discuss translational considerations, including Quality-by-Design (QbD) approaches, scalability, stability, and clinical relevance. By integrating formulation science with route-specific therapeutic goals and translational readiness, this review provides a unified framework to guide the rational development and future clinical translation of PCZ -based nanomedicine for fungal infections.
This review aims to comprehensively summarize the progress and potential of advanced nanocarrier systems for PCZ delivery, with a particular focus on topical, systemic, vaginal, and ocular applications. We will discuss the pharmacological challenges, resistance, and mode of action of PCZ, as well as the design and functionality of various nanocarriers and their therapeutic advantages in overcoming biological barriers. By consolidating recent developments, this review highlights the potential of nanotechnology-enabled delivery strategies to revolutionize antifungal treatment and pave the way for more effective, targeted, and patient-friendly formulations.
Therapeutic Challenges of Fungal InfectionsThe therapeutic landscape faces a mounting crisis due to the increasing emergence of antifungal drug resistance [33]. Particularly alarming is the rise of multidrug-resistant fungal pathogens like Candida auris, which was first identified in 2009 and has rapidly spread globally [34, 35]. C. auris exhibits intrinsic resistance to several frontline antifungals, including fluconazole, complicating treatment and outbreak control efforts [36]. The World Health Organization (WHO) has classified C. auris as a critical priority pathogen due to its resistance profile and clinical threat [37]. Other emerging resistant strains and species add complexity to managing fungal infections, exacerbated by factors such as widespread antifungal use, agricultural fungicide exposure, and environmental changes [38].
Despite the urgent need, there are several barriers that hinder effective antifungal treatment worldwide. These include diagnostic challenges caused by slow and insensitive conventional tests, misdiagnosis, and lack of routine fungal disease surveillance [39, 40]. Antifungal drugs also face limitations related to toxicity, drug-drug interactions, and poor bioavailability [41]. Furthermore, high costs and limited accessibility, particularly in resource-limited settings, prevent optimal treatment coverage [42]. The similarity between fungal pathogens and human cells makes developing new antifungal agents challenging due to potential host toxicity, slowing the pipeline of novel therapeutics [43]. Patient compliance with long-term treatments and co-morbidities also affect therapeutic outcomes adversely [44].
Antifungal Activity of PosaconazoleAspergillus species, Rhizopus species, Blastomyces dermatitidis, Coccidioides immitis/posadasii, Histoplasma capsulatum, Candida species resistant to older azoles, Cryptococcus neoformans, and other opportunistic filamentous and dimorphic fungi are among the many pathogenic fungi that PCZ exhibits broad-spectrum activity against [45]. Compared to other approved broad-range antifungal medications, PCZ covers a greater diversity of fungi. PCZ exhibits efficacy in vitro against fluconazole-resistant fungi, including Aspergillus species, Candida krusei, Candida guilliermondii, and Candida dubliniensis. Additionally, it was demonstrated that PCZ was more effective against Aspergillus species than voriconazole (VCZ) [46]. Additionally, in comparison to ITZ and VCZ, PCZ has demonstrated greater effectiveness against Aspergillus fumigatus and Aspergillus flavus [47]. In neuro-infections brought on by invasive fusariosis, zygomycosis, cryptococcal meningitis, and coccidioidomycosis, PCZ has shown activity [48,49,50]. According to Clark et al. (2015) [51] and Soysal (2015) [52], PCZ has been licensed by the Food and Drug Administration (FDA) to prevent invasive aspergillosis or Candida infections in patients with graft-versus-host disease (GVHD) undergoing hematopoietic stem cell transplantation as well as patients with hematologic malignancy who have a prolonged neutropenic period, fusariosis, or particularly mucormycosis.
ChemistryAccording to Santo, (2008) [53], PCZ is a lipophilic third-generation triazole with a unique tetrahydrofuran (THF) structure. PCZ has broad-spectrum action against a range of parasites and harmful fungi. Each of the three moieties that make up PCZ’s chemical structure, the triazolone side chain region, the linker, and the 2,2,4-trisubstituted THF unit was produced from unique critical intermediates with particular stereochemistry [54]. The triazolone ring possesses a H acceptor in N, C═O, and amines, whereas PCZ has a hydrogen bond donor in N 2 C–H (Fig. 1). Both donor and acceptor roles are played by the hydroxyl functionals. Intermolecular interactions include halogen atoms and ether bonds.
PCZ must be supplied by emulsifying in polysorbate 80, since it has a molecular weight of 700.774 g/mol [55]. PCZ, like ICZ, gives the azole target 14-α‐demethylase more sites of interaction. PCZ differs from ICZ in that it has a furan ring with fluorine and chlorine substituents [56]. PCZ shows a pka value of 3.6 and melting point of 168 °C [57]. It was also reported by Mudi et al. 2020 that PCZ has a crystalline solubility at pH 6.5 of 0.2 µg/mL, while it demonstrates a much higher crystalline solubility (719 µg/mL) at pH 1.2, indicating its superior aqueous solubility in acidic medium rather than basic one.
Fig. 1
Chemical structure of PCZ
PharmacokineticsPCZ is classified as Biopharmaceutical Classification System (BCS) Class II drug as it shows a low aqueous solubility and high permeability [58]. Three forms of PCZ are now on the market: an intravenous infusion solution, a gastro-resistant pill, and an oral suspension. Nonetheless, these formulations vary significantly in terms of pharmacokinetics, particularly during the absorption phase [59]. PCZ is very poorly soluble in water (less than 1 µg/mL) at neutral to basic pH levels, making it a poorly water-soluble drug with low and erratic oral bioavailability. Its solubility increases significantly in acidic conditions, reaching a solubility of approximately 0.8 mg/mL in the stomach (low pH) [60]. Its bioavailability is significantly enhanced when co-administered with a high-fat meal, which prolongs gastric emptying and reduces intestinal pH by stimulating gastric acid secretion [61].
Over the past decade, pharmacokinetic investigations in healthy volunteers have confirmed the impact of these factors, reporting a highly variable absolute oral bioavailability ranging from 8% to 47% for oral suspension [62]. Consequently, clinical guidelines recommend administration of PCZ with or immediately after food, particularly fatty meals, or in combination with acidic beverages to optimize systemic exposure [63]. Despite these recommendations, the relative bioavailability of the suspension formulation is approximately 55% lower in patients compared with healthy individuals [64]. Furthermore, the use of acid-suppressive therapies especially proton pump inhibitors was shown to reduce absorption and relative bioavailability by about 45% in patients with invasive aspergillosis or those at high risk of invasive fungal infection [64, 65].
Concomitant administration of metoclopramide has also been associated with a 35% decline in bioavailability. In addition, clinical conditions such as diarrhea (up to 59%), mucositis (58%), and acute GVHD in hematopoietic stem cell transplant recipients markedly compromise absorption, leading to subtherapeutic serum concentrations and treatment failure [64, 66, 67]. Conversely, dividing the daily therapeutic dose of 800 mg into two (98%) or four (220%) administrations substantially improved relative bioavailability, highlighting 800 mg as the threshold for saturable enteral absorption of the oral suspension [68].
In 2014, both the gastro-resistant tablet formulation (100 mg) and the intravenous (IV) solution (18 mg/mL) of PCZ received approval from the European Medicines Agency and the United States FDA [69, 70]. Findings from a prospective, open-label, five-way crossover trial demonstrated that administration of 400 mg of the tablet al.one produced mean AUC₀–∞, Tmax, and t½ values comparable to those observed when the same dose was co-administered with antacids, H₂ receptor antagonists, proton pump inhibitors, or metoclopramide [71]. This pharmacological profile is especially advantageous in immunosuppressed patients who frequently require acid-suppressive medications. An additional benefit of the tablet formulation is its ability to consistently achieve higher systemic concentrations, largely independent of food intake (Table 1) [78]. The IV formulation employs sulfobutylether-β-cyclodextrin as a solubilizing agent; however, because it can irritate the venous wall, administration via a central venous catheter is recommended [79, 80].
A retrospective analysis involving critically ill individuals and transplant recipients reported that the IV preparation was approximately 20-fold more likely, and the gastro-resistant tablet 7.7-fold more likely, to achieve therapeutic plasma concentrations compared with the oral suspension [81]. PCZ exhibits extensive tissue distribution, with a large apparent steady-state volume of distribution (Vd, ss) [65, 82] which varies by formulation: 30–70 L/kg with the oral suspension, 5–6 L/kg with the delayed release tablet, and 3–4 L/kg with the IV solution [83]. The distribution volume is further increased in patients with febrile neutropenia or refractory invasive fungal infections [73]. Protein binding is high (98–99%), non–concentration dependent, and primarily to plasma proteins [84]. Notably, drug accumulation in pulmonary alveolar cells greatly exceeds plasma levels, with cell-to-plasma ratios ranging from 27.3 ± 18.0 to 44.3 ± 44.2 in healthy individuals [85]. Furthermore, serum concentrations correlate strongly with levels detected in bronchoalveolar lavage fluid of lung transplant recipients [86].
PCZ undergoes biotransformation primarily through glucuronidation, producing multiple inactive glucuronide conjugates without the need for prior cytochrome P450 (CYP)-mediated oxidation. Hydroxylated and oxidized derivatives account for only about 2% of its metabolites, with uridine 5′-diphospho-glucuronosyltransferase (UGT) isoform UGT1A4 identified as the main hepatic enzyme catalyzing this process [87, 88]. Renal elimination plays only a minor role in drug clearance, as approximately 14% of radiolabeled PCZ recovered in the urine of healthy volunteers was detected almost exclusively as inactive mono- and diglucuronide conjugates. In contrast, the predominant route of excretion is via feces (77%), primarily in the unchanged form of the parent compound [89]. Following a single oral dose, the drug exhibits an apparent clearance of roughly 16.4 L/h and a plasma elimination half-life ranging from 25 to 34 h [90].
Table 1 Pharmacokinetic parameters of PCZMechanism of ActionCYP51 is a pivotal enzyme for the biological synthesis of sterols in fungal cellular membranes [91]. CYP51 catalyzes the removal of the 14-methyl group in lanosterol through a series of sequential oxidative reactions. Ultimately, 14-demethyl lanosterol is transformed into ergosterol, which constitutes a significant component of the fungal cellular membrane. Triazole antifungal agents can impede CYP51 and sterol 14α-demethylation. PCZ, akin to other triazoles, induces the accumulation of 14α-methylated sterols, thereby disrupting the composition and functionality of the cellular membrane [92, 93]. It appears that PCZ exhibits a more favorable activity profile in relation to fluconazole (FLZ) concerning the inhibition of CYP51 across numerous yeast strains. Furthermore, PCZ demonstrates efficacy against the mutated variant of CYP51 in C. albicans, which is resistant to FLZ, ITZ, and VCZ [94, 95].
Epidemiology of Azole ResistanceAzole drugs, such as fluconazole, VCZ, and PCZ, target the cytochrome P450 enzyme sterol 14α-demethylase, which is encoded by Cyp51 in molds and ERG11 in yeast and converts lanosterol to ergosterol [96]. According to Singh et al. (2023) [97], inhibition of 14α-demethylase exhibits fungicidal effects on molds and fungistatic effects on yeasts. According to Lewis et al. (2024) [98], triazoles are often used to treat candidiasis and are advised for the treatment of aspergillosis. According to epidemiological research, Aspergillus and Candida species have significant azole resistance [99], but Cryptococcus species continue to exhibit modest levels of azole resistance [100]. In general, acquired resistance and an epidemiological shift toward species that are naturally less sensitive have led to the emergence of drug resistance [35]. It is generally known that C. krusei has inherent azole resistance and that Candida glabrata has high rates of azole re
Comments (0)