
Remember me
Inflammation is an immune-mediated, nonspecific host response to external stimuli or tissue injury. It involves immune cells, soluble molecular mediators, and the microvascular network, and represents one of the principal mechanisms by which the host eradicates pathogens, repairs damaged tissue, removes cellular debris, and re-establishes homeostasis. Both endogenous triggers such as ischemia or atherosclerosis and exogenous triggers such as microbial infection can initiate the response. The immune system normally maintains a balance between pro- and anti-inflammatory mediators; failure of anti-inflammatory signals to restrain pro-inflammatory pathways can lead to persistent inflammation. Aging dysregulates both the innate and adaptive immune compartments, producing a state of chronic, low-grade inflammation 1.
Inflammation may be precipitated by thrombotic ischemic stroke, immune dysregulation, malignancy, exposure to chemicals such as tobacco smoke, dioxins, or polycyclic aromatic hydrocarbons; by physical insults including hemorrhagic stroke or trauma; and by neurological conditions such as Alzheimer’s disease and depression. In addition, infections caused by bacterial, fungal, viral, or protozoan pathogens invariably evoke inflammatory responses 2.
Inflammation represents a common pathogenic denominator across chronic diseases. Its involvement is well established in endocrine disorders, autoimmune conditions such as rheumatoid arthritis, gout, inflammatory bowel disease, and in hypersensitivity reactions such as anaphylaxis, as well as in environmentally induced diseases that follow asbestos exposure or smoke inhalation. Metabolic disorders, notably diabetes mellitus, also exhibit an inflammatory milieu. Over the past three decades, evidence has accumulated that an even broader spectrum of diseases—including myocardial ischemia, acute cerebrovascular events, chronic Alzheimer’s disease, and chronic arterial or venous insufficiency—display inflammatory signatures at the cellular and molecular levels 3.
Activation of immune and parenchymal cells eradicates pathogens and promotes tissue repair, thereby protecting the host from viruses, bacteria, toxins, and other insults. Across evolution, inflammation has been accompanied by metabolic and neuroendocrine adaptations that conserve energy and allocate substrates to the immune system, with the specific pattern depending on the intensity and systemic spread of the response. The resultant “sickness behaviours” constitute energy-saving strategies integral to inflammatory biology, underscoring the robustness of this defence system 4.
Microcirculatory phenomena exemplify these principles. The tissue site, the affected organ, and the nature and severity of injury—whether mechanical, infectious, chemical, thermal, radiogenic, or ischemic—critically determine the qualitative features of the ensuing inflammatory response 4.
Isolated endothelial injuryA highly focused laser burn provides a precise experimental model for interrogating inflammation at near-single-cell resolution. Laser-induced microvascular endothelial damage results in rapid platelet adhesion to the denuded surface and to each other, with minimal entrapment of erythrocytes; a platelet thrombus therefore forms swiftly at the injury site. Depending on lesion size, the thrombus may stabilise and subsequently detach, or it may expand to occlude the microvessel, whereupon detachment can lead to fragmentation 4.
Parenchymal burn injuryWhen the laser is applied to a confined tissue region encompassing multiple cell layers, a different cascade unfolds. Neighbouring microvessels—even if not directly exposed to the laser—exhibit increased permeability. Circulating leukocytes adhere to the intact endothelium of upstream vessels and subsequently migrate chemotactically toward the burn locus 4.
Shock physiologyCirculatory shock—whether precipitated by massive haemorrhage, extensive burns, or severe trauma—elicits a distinct systemic inflammatory cascade. Organs spared from the primary insult may nonetheless be damaged by downstream inflammatory mediators; the intestine and pancreas are major sources of such effector signals. Intestinal inflammation can propagate systemically and injure remote organs, including vascular beds such as the superior mesenteric artery that were not part of the initiating event 3.
This review summarises current knowledge on inflammation and anti-inflammatory pharmacotherapy, with the goal of optimising clinical outcomes in inflammation-associated disorders. It discusses biomarkers of acute and chronic inflammation, their roles in pathogenesis, established and emerging therapeutic strategies, and prospective molecular targets.
Acute InflammationInjury-induced release of pro-inflammatory cytokines can escape the local milieu and enter the circulation, generating a “cytokine storm”. Tumour necrosis factor-α is a pivotal component of this loop. Systemic cytokine signalling activates organ-specific receptors, eliciting a coordinated acute-phase response, most prominently in the liver. Hepatocytes up-regulate acute-phase proteins such as lipopolysaccharide-binding protein, complement component 3, haptoglobin, serum amyloid A, C-reactive protein, fibrinogen, and ceruloplasmin, while down-regulating cortisol-binding globulin, zinc and iron transporters, albumin, transferrin, transthyretin, and retinol-binding protein. Activation of the renin–angiotensin–aldosterone system further sustains low-grade inflammation via angiotensin II and aldosterone, which drive oxidative stress, endothelial dysfunction, and immune-cell recruitment 5.
Chronic InflammationInflammation that persists beyond its physiological purpose or arises without a discernible stimulus is defined as chronic. It commonly results from unresolved infections, either because pathogens evade immunity or because persistent antigens overwhelm host defences; however, refractory chemical or physical insults and genetic predisposition also contribute. Transition from acute to chronic inflammation is orchestrated by sustained production of cytokines (e.g., TNF-α, IL-1β, IL-6, CXCL8), continuous activation of macrophages, neutrophils, and T cells, defective resolution programmes, tissue remodelling driven by transforming growth factor-β, and altered cell-death pathways. Chronic inflammation perturbs metabolism, inducing insulin resistance, adipose accumulation, and dyslipidaemia; it simultaneously promotes systemic oxidative stress, mitochondrial dysfunction, and a perpetuating cytokine milieu, underpinning disorders such as obesity, type 2 diabetes, cardiovascular disease, and neurodegeneration. In the central nervous system, persistent inflammation fosters protein aggregation and compromises the blood–brain barrier, exacerbating neuronal injury and neurodegenerative diseases 6.
ROLE OF INFLAMMATION IN DISEASESAutoimmune Diseases and InflammationTargeting inflammatory mediators and their signalling pathways represents a promising therapeutic strategy for autoimmune diseases (AIDs). Inflammasomes and other inflammatory mediators play a central role in AID pathophysiology by modulating B- and T-cell responses within both the innate and adaptive immune compartments and ultimately driving autoimmune reactivity. This review therefore summarises the contribution of inflammation to various AIDs and outlines current or investigational inhibitors directed at key inflammatory components 20.
It has long been recognised that Th1, Th2 and Th17 subsets, as well as other T-cell-mediated responses, are critical to the initiation of autoimmune disorders. Compelling evidence now indicates that dysregulated Th1, Th2 and Th17 responses make a major contribution to autoimmune inflammation. Vasoactive intestinal peptide (VIP) has recently been shown to modulate the effector functions of several cell populations involved in rheumatoid arthritis, including macrophages, fibroblast-like synoviocytes and lymphocytes 21.
Chronic Diseases and InflammationInflammation is a pivotal factor in the development of atherosclerosis and, consequently, cardiovascular disease. Tobacco smoking is a well-established risk factor for this multifactorial disorder. After adjustment for age, region and sex, the INTERHEART study (12,438 cases; 14,605 controls) showed that current smokers had a markedly higher risk of myocardial infarction (odds ratio = 2.3; 95 % CI = 2.1–2.6) than never-smokers 22.
The inflammatory state associated with metabolic syndrome is unusual, as it occurs without overt infection, autoimmunity or marked tissue damage. Because the magnitude of activation is modest, it is commonly referred to as ‘low-grade’ chronic inflammation. To describe an intermediate state between basal and overt inflammation, some authors have coined the terms ‘metaflammation’ (metabolism-induced inflammation) and ‘parainflammation’. Regardless of nomenclature, the inflammatory milieu that characterises metabolic syndrome displays unique features whose mechanisms remain incompletely understood 23.
Cancer and InflammationTumour development is accompanied by the expansion of stroma, vasculature, infiltrating inflammatory cells and malignant cells; crosstalk among these compartments fuels tumour progression. Early observations indicated that acute inflammation can suppress or eradicate nascent malignancies 24.
First proposed by Rudolf Virchow more than a century ago, the link between inflammation and cancer has since been corroborated by genetic and molecular studies. Epidemiological, clinical and experimental data demonstrate that persistent inflammation predisposes individuals to specific cancers and accelerates tumourigenesis. Hallmarks of cancer-related inflammation include expression of tumour necrosis factor (TNF) and interleukin-1 (IL-1), leukocyte infiltration, chemokine production (e.g. CCL2, CXCL8), dynamic tissue remodelling and neovascularisation 25.
A tumour can be viewed as a chronic, non-healing inflammatory wound. Primary tumour sites harbour a spectrum of inflammatory cells—from myeloid and lymphoid lineages and from innate and adaptive immunity—in addition to the tumour cells themselves. These cells influence diverse tumour phenotypes and biological behaviours. Three principal myeloid subsets infiltrate tumours: tumour-associated neutrophils (TANs), tumour-associated macrophages (TAMs) and Gr-1⁺CD11b⁺ immature myeloid cells. Gr-1⁺CD11b⁺ cells constitute a heterogeneous population, whereas TAMs and TANs are more differentiated. The phenotypic and functional interrelationships of these three subsets within the tumour micro-environment (TME) remain to be fully elucidated. Because of their potent immunosuppressive activity, Gr-1⁺CD11b⁺ cells are generally termed myeloid-derived suppressor cells (MDSCs) 26.
Therapeutic Strategies in InflammationC-reactive protein (CRP), erythrocyte sedimentation rate (ESR), interleukin (IL)-1β, IL-6, tumour necrosis factor-α (TNF-α), matrix metalloproteinases (MMPs), fibrinogen, myeloperoxidase and leukotrienes constitute a recognised panel of inflammatory biomarkers that support both diagnosis and longitudinal monitoring. Resolution of inflammation is an essential component of tissue repair and involves tightly coordinated events, including clearance of inflammatory cells and mediators, restoration of vascular integrity, activation of anti-inflammatory signalling and phagocytosis, stimulation of tissue regeneration, transition to a reparative phenotype and prevention of chronic inflammation. Among the available interventions, non-steroidal anti-inflammatory drugs (NSAIDs) remain the most widely used; approximately 30 million people worldwide take these agents daily. NSAIDs are generally effective against acute and chronic inflammatory conditions. More than 40 individual NSAIDs are marketed and are typically classified by chemical structure and anticipated risk profile. In general, NSAIDs display small volumes of distribution, high plasma-protein binding, limited first-pass hepatic metabolism and good oral absorption. Routes of administration, elimination half-lives and tolerability vary among agents, although members of the same chemical class usually share similar pharmacokinetic characteristics. Globally, ibuprofen and diclofenac are the most frequently used NSAIDs, followed by naproxen, indomethacin, piroxicam and ketoprofen. Aspirin, naproxen and ibuprofen are available over the counter for common indications such as headache and postoperative pain, whereas the majority of NSAID prescriptions are generated in primary care settings 27.
Non-Steroidal Anti-Inflammatory DrugsIn addition to their analgesic, anti-inflammatory and antipyretic properties, NSAIDs inhibit platelet aggregation 28. Their potential antidepressant effects are under active investigation; consequently, this discussion is divided into ‘selective COX-2 inhibitors’ and ‘non-selective COX inhibitors’, as selective COX-2 blockade is hypothesised to confer superior anti-inflammatory—and thus antidepressant—activity 29. NSAIDs exert their principal effects by inhibiting prostaglandin synthesis through cyclo-oxygenase (COX)-1 and COX-2 blockade. They are therefore categorised into traditional non-selective agents, which inhibit both isoforms, and newer selective COX-2 inhibitors that preferentially target COX-2, although pharmacodynamic overlap exists 30.
COX-1 is constitutively expressed and fulfils several physiological roles; activation in gastric mucosa and vascular endothelium generates cytoprotective and antithrombogenic prostacyclin. In contrast, COX-2, characterised in the early 1990s, is inducible in response to pro-inflammatory stimuli in multiple cell types. Needleman et al. first postulated its existence after observing lipopolysaccharide-induced prostaglandin production in mouse peritoneal macrophages and human monocytes—an effect suppressed by dexamethasone and associated with de novo COX protein synthesis 31.
Mechanism of Action of NSAIDsSynovial fluid from patients with rheumatoid arthritis contains ≈20 ng ml⁻¹ of prostaglandin E₂ (PGE₂); this level falls to undetectable concentrations after aspirin therapy, indicating clinically significant suppression of prostaglandin synthesis. In a rat model of carrageenan-induced inflammation, subcutaneous implantation of polyester sponges led to a steady increase in PGE₂ within the first 24 h, whereas thromboxane A₂ (TXA₂) and leukotriene B₄ (LTB₄) peaked at 4–6 h and declined thereafter. PGE₂ mediates vasodilatation and hyperalgesia, while LTB₄ recruits polymorphonuclear leukocytes; the role of TXA₂ remains unclear 32.
Prostaglandins are lipid mediators derived from arachidonic acid via the COX pathway and participate in numerous physiological and pathological processes, including pain, inflammation, fever, oncogenesis and neurological disease. Arachidonic acid, a 20-carbon polyunsaturated fatty acid esterified within membrane phospholipids, is liberated by phospholipase A₂ and converted by COX or lipoxygenase enzymes into eicosanoids. COX-1 and COX-2 catalyse the first committed step in prostaglandin and thromboxane biosynthesis 33.
This review included randomised controlled trials of NSAIDs in which pain was an outcome for acute conditions (e.g., strains, sprains, sports injuries) or chronic disorders (e.g., arthritis, rheumatism). Trials involving thrombophlebitis, experimental pain models, vaginitis or oral/buccal conditions were excluded 34.
A summary of NSAID classes, mechanisms of action and clinical implications is provided in Table 1.
Table 1Summary of NSAID classes, mechanisms and clinical implications.
ClassExamplesMechanismClinical UsesKey Side EffectsContra-indicationsSalicylatesAspirinIrreversible COX-1 and COX-2 inhibitionPain, fever, antiplatelet (e.g., for cardiovascular protection), anti-inflammatoryGI bleeding, Reye's syndrome (in children with viral infections), tinnitus, hypersensitivity reactionsChildren with viral infections, pregnancy (3rd trimester), bleeding disorders, hypersensitivity to salicylatesPropionic acid derivativesIbuprofen, Naproxen, KetoprofenReversible COX-1 and COX-2 inhibitionPain, inflammation, feverGI upset (dyspepsia, ulcers), CV risk (dose-dependent, e.g., MI, stroke), renal impairmentPeptic ulcer disease, CV disease, renal impairment, hypersensitivityAcetic acid derivativesDiclofenac, IndomethacinReversible COX-1 and COX-2 inhibitionPain, inflammation, gout (indomethacin), patent ductus arteriosus (indomethacin)High GI risk (ulcers, bleeding), CV risk (e.g., MI, stroke), CNS effects (e.g., headache, dizziness), renal impairmentPeptic ulcer disease, CV disease, renal impairment, hypersensitivityEnolic acid derivatives (Oxicams)Piroxicam, MeloxicamReversible COX-1 and COX-2 inhibition (meloxicam: some COX-2 selectivity)Pain, inflammationGI upset, CV risk (e.g., MI, stroke), photosensitivity (piroxicam)Peptic ulcer disease, CV disease, renal impairment, hypersensitivityFenamic acid derivativesMefenamic acidReversible COX-1 and COX-2 inhibitionPain, inflammationGI upset, diarrhoea, haemolytic anaemia (rare)Peptic ulcer disease, renal impairment, hypersensitivitySelective COX-2 inhibitorsCelecoxib, EtoricoxibSelective COX-2 inhibitionPain, inflammation, arthritis (lower GI risk vs. non-selective NSAIDs)Increased CV risk (e.g., MI, stroke), fluid retention, oedemaHistory of CV disease, sulphonamide allergy (celecoxib), hypersensitivityNSAIDs and corticosteroidsBecause of their anti-inflammatory and analgesic properties, non-steroidal anti-inflammatory drugs (NSAIDs) are useful in the management of rheumatoid arthritis (RA). In many patients, these agents may adequately control symptoms and obviate the need for disease-modifying antirheumatic drugs (DMARDs). Furthermore, patients with suspected RA often receive NSAIDs to provide temporary symptom relief until a definitive diagnosis is established or disease progression warrants initiation of DMARD therapy 35.
NSAIDs have been shown to be effective in treating acute pain, thereby representing one of the first-line pharmacological treatments for arthritic pain. However, expert opinions and current guidelines differ regarding whether NSAIDs are superior to paracetamol as first-line analgesic therapy for arthritic disorders. In a recent meta-analysis of 15 RCTs including 5,986 participants, NSAIDs were significantly more effective than paracetamol for knee and hip pain in osteoarthritis, although the effect size for both therapies was modest. NSAIDs are also frequently used to treat RA symptoms, but the effects are likewise limited 36.
NSAIDs and cancerAccumulating evidence over several decades has demonstrated the anti-neoplastic activity of NSAIDs. After the identification of the COX-2 isoform, research revealed that it is highly expressed in several malignancies—including those of the breast, colon, pancreas, and prostate—and appears to regulate numerous cellular functions. Because of their potential role in cancer prevention and therapy, selective COX-2 inhibitors have been the subject of intense investigation. Both COX-dependent and COX-independent mechanisms are believed to contribute to the anticancer effects of COX-2 inhibitors and other NSAIDs. COX-2 is highly inducible in response to growth factors, tumor promoters, and inflammatory cytokines, whereas COX-1 is constitutively expressed in most tissues and fulfils a “housekeeping” role 33.
NSAIDs and COVID-19The relevance of these findings to the COVID-19 pandemic remains uncertain. Data on intermittent NSAID use are limited to primary-care trials that evaluated short-term dosing during respiratory infections. Nevertheless, sporadic NSAID administration may help patients with COVID-19 by relieving nocturnal symptoms and promoting sleep when paracetamol is inadequate—sleep being important for immune defence. The available evidence does not support routinely advising against NSAID use. Early concerns that antihypertensives or ibuprofen might exacerbate COVID-19 severity have not been substantiated. Moreover, NSAIDs may be required to treat co-existing symptoms such as musculoskeletal pain in patients with COVID-19 37.
NSAIDs: COX-independent anti-inflammatory effectsThe clinical response to NSAIDs varies markedly between individuals, suggesting differences beyond prostaglandin inhibition. Pharmacokinetic factors such as enantiomeric configuration, serum concentrations, or metabolism do not fully explain this variability. These observations imply that NSAID-induced, prostaglandin-independent pathways play an important role in determining individual responsiveness 38.
Non-selective NSAIDs and inflammationNaproxen is a non-steroidal anti-inflammatory drug with analgesic, anti-inflammatory, and antipyretic properties attributable to inhibition of prostaglandin synthesis. It is a widely used over-the-counter (OTC) agent with favourable efficacy and a comparatively low adverse-effect profile. Naproxen is chiral, existing as the (S)- and (R)-enantiomers; the (S)-form is approximately 28-fold more potent than the (R)-form in anti-inflammatory and analgesic activity 39.
Ibuprofen is among the most commonly used analgesic, antipyretic, and anti-inflammatory OTC medications worldwide. Usage patterns differ by country, but it is generally ranked behind aspirin and paracetamol for self-medication of fever, inflammation, and acute pain. Of these three agents, OTC ibuprofen is considered the safest, being rarely associated with fatal or severe adverse events. Consequently, it has been referred to as the “mildest” NSAID in long-term clinical use 40.
Oxaprozin, a propionic-acid derivative, is an achiral oxazole with moderate lipophilicity and a pKa of 6.1 in water. These physicochemical properties may underlie its favourable gastric tolerability. Furthermore, its distribution within lipophilic media between pH 2 and 4 varies less than that of many NSAIDs, suggesting slower gastric cellular uptake and potentially reduced mucosal injury 41.
Piroxicam is a non-steroidal anti-inflammatory drug that inhibits platelet aggregation and possesses analgesic, antipyretic, and anti-inflammatory activity. It has an elimination half-life of approximately 38 h and is primarily cleared by hepatic metabolism to inactive metabolites. Clinical trials have demonstrated the efficacy of piroxicam in osteoarthritis and rheumatoid arthritis; smaller studies also support its use for acute musculoskeletal disorders and gouty arthritis 30.
Structures of naproxen, ibuprofen, oxaprozin, and piroxicam are presented in Figure 1.
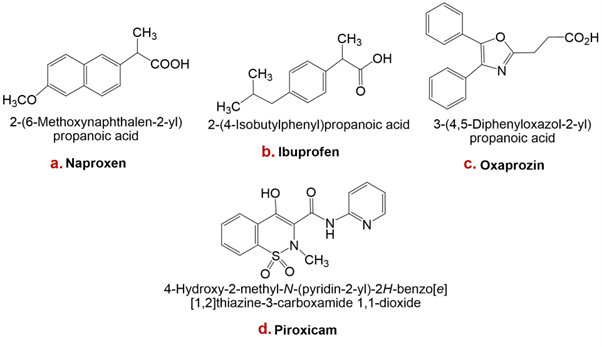 Figure 1
Figure 1Structures of some non-steroidal anti-inflammatory drugs (NSAIDs): a. Naproxen, b. Ibuprofen, c. Oxaprozin, d. Piroxicam.
COX-2 selective NSAIDs and InflammationCelecoxib is a selective cyclooxygenase-2 (COX-2) inhibitor that exhibits anti-inflammatory, antipyretic, and analgesic activity. It is approved for the treatment of acute pain, ankylosing spondylitis, rheumatoid arthritis, and osteoarthritis. In patients with familial adenomatous polyposis (FAP), celecoxib is used as an adjuvant to surgery to reduce the number of adenomatous colorectal polyps and has also shown potential chemopreventive effects in other malignancies. Celecoxib’s ability to reduce inflammation and discomfort stems from its specific suppression of prostaglandin G/H synthase-2 (encoded by gene PTGS2), thereby decreasing prostaglandin synthesis. PTGS exists as two isoenzymes, PTGS1 and PTGS2, both possessing cyclooxygenase and hydroperoxidase activities 42.
Valdecoxib is another NSAID indicated for primary dysmenorrhoea as well as for the symptomatic treatment of osteoarthritis and rheumatoid arthritis. It is a potent and highly selective COX-2 inhibitor; because COX-2 is an inducible isoenzyme that is principally expressed in inflamed tissues, inhibition by valdecoxib spares constitutive COX-1 activity in most physiological tissues 43. Drugs in this class, also referred to as coxibs, COX-2-specific inhibitors, COX-2-selective NSAIDs or COX-1-sparing NSAIDs, exhibit high potency and slow dissociation from the COX-2 active site, a property conferred by their extended side chains that occupy the hydrophobic channel of the enzyme. At therapeutic concentrations valdecoxib does not inhibit COX-1 in humans (43, 44). Although it is associated with a lower incidence of peptic ulceration and gastrointestinal bleeding than non-selective NSAIDs, valdecoxib confers a higher risk of serious cardiovascular events and was withdrawn from the market in 2005 because of an excess incidence of myocardial infarction and stroke at high doses or after prolonged use. Representative chemical structures of celecoxib and valdecoxib are shown in Figure 2.
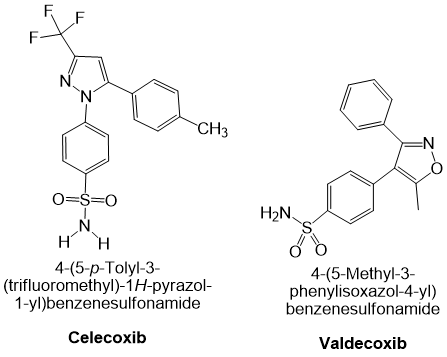 Figure 2
Figure 2Cyclooxygenase-2 (COX-2) selective non-steroidal anti-inflammatory drugs (NSAIDs): Chemical structures of celecoxib and valdecoxib.
Chronic administration of NSAIDs can result in adverse effects including peptic ulcer disease, gastrointestinal bleeding, renal impairment, hepatotoxicity and cardiovascular events. While these drugs are efficacious for the management of acute pain and inflammation, they do not abrogate the underlying pathophysiological processes of chronic inflammatory disorders such as rheumatoid arthritis and inflammatory bowel disease. Consequently, there is a pressing need to identify novel anti-inflammatory agents. Lifestyle modifications provide a complementary strategy with fewer and less severe adverse effects, and may reduce pharmacological requirements and their attendant risks 44,45.
Lifestyle Modification and InflammationRole of diet in inflammation: Most individuals in Western societies adopt lifestyles that markedly increase their risk of developing chronic metabolic disorders, including cancer, diabetes, neurodegenerative diseases, and cardiovascular disease. Chronic low-grade inflammation accompanying these conditions is mediated by activation of several molecular pathways, notably NF-κB, MMP-9 (matrix metalloproteinase-9), MAPKs (mitogen-activated protein kinases), COX-2 (cyclo-oxygenase-2), and STAT-3 (signal transducer and activator of transcription-3). Matrix metalloproteinases (MMPs) degrade the extracellular matrix, facilitating immune-cell infiltration and perpetuating inflammation. Numerous intervention studies demonstrate that lifestyle modification can attenuate inflammation and improve health outcomes. Fermentable dietary fibres escape enzymatic digestion in the small intestine, reach the colon, and are converted by the gut microbiota into the short-chain fatty acids (SCFAs) acetate, propionate, and butyrate. These SCFAs are absorbed systemically and suppress inflammatory signalling; conversely, germ-free animals, which lack tissue and circulating SCFAs, display exaggerated inflammatory flare-ups 45.
Fruits and vegetables should constitute a major component of an anti-inflammatory diet. They are energy-sparse yet rich in vitamins, minerals, and phytochemicals, and should be consumed in large quantities, in a variety of colours and types, at every meal. Their high polyphenol content underlies both their vivid pigmentation and their anti-inflammatory activity 46.
Omega-6 fatty acids serve as precursors of many pro-inflammatory eicosanoids; however, arachidonic acid, not linoleic acid (the predominant dietary omega-6 fatty acid), is the immediate substrate for eicosanoid synthesis. The rate-limiting desaturases Δ6- and Δ5-desaturase regulate conversion of linoleic acid to arachidonic acid and are modulated by hormonal and nutritional factors. A principal aim of anti-inflammatory diets is therefore to dampen systemic inflammation in conditions such as arthritis, cardiovascular disease, and neurodegenerative disorders 47.
Exercise and inflammatory markers: Cross-sectional studies reveal an association between physical inactivity and low-grade systemic inflammation in healthy older adults and patients with intermittent claudication, although causality cannot be inferred. Two long-term trials showed that regular exercise reduces C-reactive protein (CRP) concentrations, supporting the hypothesis that habitual physical activity mitigates systemic inflammation. In a controlled study, healthy volunteers received a low dose of Escherichia coli endotoxin to induce experimental low-grade inflammation; participants randomised to exercise before endotoxin administration exhibited significantly lower tumour necrosis factor (TNF-α) responses than those at rest 48.
Paradoxically, the transient exercise-induced rise in pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) triggers subsequent production of anti-inflammatory mediators, reactive oxygen and nitrogen species (O2−, NO), and manganese superoxide dismutase (Mn-SOD). This cascade culminates in release of transforming growth factor-β (TGF-β), IL-10, and IL-1 receptor antagonist. Endurance exercise elicits a greater cytokine response than moderate activities such as walking, cycling, tennis, or rowing; skeletal muscle and peripheral blood mononuclear cells are the principal sources of these cytokines 49.
The optimal exercise prescription for reducing inflammation is still being elucidated. Compared with habitual physical activity, structured exercise exhibits a dose-dependent relationship with inflammatory mediators and health outcomes. Emerging research on sedentary behaviour and light-intensity activity further emphasises their associations with vascular dysfunction and cardiovascular risk. Current evidence supports regular moderate-intensity aerobic exercise (30–60 min per session, 3–5 days per week), which reduces visceral adiposity, improves insulin sensitivity, lowers oxidative stress, increases anti-inflammatory cytokines (e.g., IL-10), and modulates immune-cell profiles (higher regulatory T-cell and lower macrophage counts), thereby attenuating systemic inflammation 50.
Mechanism of action of phenolic compounds as medications that reduce inflammationPhenolic compounds inhibit pro-inflammatory mediators other than cyclo-oxygenase (COX) by decreasing their production or activity, a mechanism that parallels that of non-steroidal anti-inflammatory drugs (NSAIDs). Furthermore, several phenolic agents can both up- and down-regulate transcription factors, such as nuclear factor-κB (NF-κB). Dietary polyphenols attenuate inflammation through diverse mechanisms, including antioxidant activity, inhibition of pro-inflammatory signaling pathways, modulation of cytokine release, and regulation of immune responses. The chemical structure of phenolic compounds critically influences their anti-inflammatory mechanisms. For example, when the C ring is unsaturated, resonance stabilizes the intermediate radical species. Moreover, a C2–C3 double bond promotes coplanarization of rings A and C, enhancing the flavonoid’s interaction with the enzyme’s active site. The catechol group on the B ring facilitates enzymatic oxidation, generating electrophilic species that permit subsequent nucleophilic addition 51.
Emerging Therapies of InflammationNovel targets of inflammationThere are five known mammalian inflammatory caspases, and all possess an N-terminal caspase-activation-and-recruitment domain (CARD). On chromosome 11q22, human inflammatory caspases are arranged as follows: caspase-12, caspase-4, caspase-5, and caspase-1. In mice, the syntenic region is arranged similarly, with caspase-12, caspase-11 and caspase-1. Comparable to murine caspase-11, human caspase-4 and ‑5 are duplicates, according to phylogenetic analysis. Moreover, the tissue-distribution patterns of caspase-11 and caspase-4 mRNA are comparable, and lipopolysaccharide (LPS) and interferon (IFN) induce the expression of both caspase-5 and caspase-11. Genes generated by duplication of the caspase-1 locus are located immediately downstream in the human genome. The three caspase-1 inhibitors encoded by these gene products—ICEBERG, INCA and COP—all contain a single CARD domain. Small molecules such as VX-765, a caspase-1 inhibitor, have been tested in clinical trials for the inflammatory disease rheumatoid arthritis. In IBD and related conditions, agents such as apremilast and canakinumab (which primarily targets IL-1β) have shown therapeutic potential. Epigenetic mechanisms, including DNA methylation, histone modifications and non-coding RNA regulation, modulate the activation or repression of inflammation-related genes (e.g., NF-κB targets) in response to infection, stress and environmental stimuli; consequently, they constitute another promising therapeutic avenue. Therefore, these mechanisms hold promise for the development of targeted therapies for inflammatory diseases. miR-155 and miR-21 enhance the inflammatory response by promoting cytokine expression and activating NF-κB. miR-146a helps terminate excessive inflammation by suppressing NF-κB and other pro-inflammatory pathways. Consequently, miRNAs are currently being explored as potential therapeutic targets in inflammatory diseases 52.
Inflammation in CKDNumerous mediators are involved in the intricate chain of events that constitutes the inflammatory response, which affects multiple cell types. Measuring one or more of these mediators can help determine the presence of inflammation. This can be achieved by assessing easily accessible and inexpensive markers such as the white-blood-cell count or serum albumin levels. Unfortunately, because they can be influenced by many other conditions, these markers are often non-specific. More precise indicators of inflammation, such as C-reactive protein and IL-6, provide a more detailed assessment but are costlier and not universally available in routine practice 53.
Inflammation in atherosclerosisDespite therapeutic advances, atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of morbidity and mortality worldwide. Inflammation is pivotal to atherogenesis and can precipitate plaque rupture and acute coronary syndrome. Although statins markedly reduce cardiovascular events in both primary and secondary prevention, many patients still experience recurrent adverse events. Therefore, targeting inflammatory pathways represents a promising strategy for novel atherosclerosis therapeutics. RAAS activity is crucial in the pathogenesis of many cardiovascular diseases. Targeting RAAS components with inhibitors such as ACE inhibitors, angiotensin-receptor blockers and aldosterone antagonists can mitigate these pro-inflammatory effects and improve outcomes. All of the above strategies can likewise be applied as emerging approaches to target chronic inflammation 54.
Precision medicine for inflammationThere is evidence of a correlation between circulating IL-6 concentrations, serum C-reactive protein (CRP) levels, and serum amyloid A (SAA) concentrations. More individualized immunotherapies may be possible if inflammatory biomarkers are dynamically monitored to determine vaccination risks and to tailor therapy. These therapies could target cytokines such as IL-1, TNF-α, or IL-6 to reduce inflammation, or employ immune adjuvants to augment the immune response. IL-6 is crucial for the differentiation of dendritic cells (DCs), maintaining them in an immature, tolerance-associated state. The inflammatory response—including DC maturation—is initiated early during infection and subsequently amplified. Biologics such as anti-TNF agents have revolutionized treatment, but not all patients respond. Side effects and resistance may also occur in some patients 55. Baseline systemic inflammatory status predicts the clinical outcome under treatments such as immune checkpoint inhibitors, anticancer vaccines, or combination therapies. However, because of methodological heterogeneity, it is challenging to determine the predictive significance of IL-6/CRP across studies; values may lie below the median or cluster into distinct subgroups. Overall, a better prognosis is associated with lower CRP levels. In the tumor microenvironment, IL-6-mediated STAT3 activation inhibits functional DC maturation, thereby reducing effector T-cell activation and impairing anticancer immunity 56. IL-6 signalling exhibits both pro- and anti-inflammatory properties: it initially promotes the differentiation of macrophages and T- and B-lymphocytes, and later contributes to the resolution of inflammation 57. Cis-signalling is restricted to cells that express IL-6Rα, including hematopoietic and hepatic cells. By contrast, IL-6 trans-signalling supports anti-tumour adaptive immunity by directing lymphocytes to tumour sites and draining lymph nodes. Consequently, precise modulation of inflammation may be more important than merely reducing toxicity, particularly during immunotherapy. Comprehensive biomonitoring should include quantification of soluble IL-6 receptors and assessment of CRP conformational isoforms (monomeric versus pentameric) when evaluating anti-IL-6 or anti-IL-6R therapies 58. The clinical implementation of precision medicine still faces several hurdles, including high biomarker costs, prolonged turnaround times, and limited accessibility. Identifying suitable biomarkers often requires expensive testing, making access difficult for many patients, especially those in rural areas. These high costs also hinder widespread insurance coverage.
NLRP3 InhibitorsNumerous cell types, including neutrophils, macrophages, microglia, lymphocytes, epithelial cells, osteoblasts, neurons and dendritic cells, express the 118-kDa cytosolic pattern-recognition receptor (PRR) NLRP3. The NLRP3 protein comprises an N-terminal pyrin (PYD) domain that recruits proteins for inflammasome assembly, a central ATPase-containing NACHT (found in NAIP, CIITA, HET-E and TP1) domain that promotes oligomerisation, and a C-terminal leucine-rich repeat (LRR) domain. Similar to other inflammasomes, the NLRP3 inflammasome consists of a sensor (NLRP3), an adaptor [apoptosis-associated speck-like protein containing a CARD (ASC)], and an effector (caspase-1). Formation of the NLRP3 inflammasome proceeds in two steps: priming and activation. During priming, stimulation with pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) such as lipopolysaccharide (LPS), or with cytokines including TNF and IL-1β, engages TLR, NOD2, IL-1R or TNF receptor signalling pathways, leading to transcriptional up-regulation of NLRP3 and of the pro-forms of the inflammatory cytokines IL-1β and IL-18 59. Recognition of PAMPs and DAMPs activates signalling proteins and transcription factors, including activator protein-1 (AP-1), nuclear factor-κB (NF-κB) and myeloid differentiation primary response protein 88 (MyD88), thereby amplifying NLRP3 and cytokine expression. Although priming has long been considered to affect only transcription and protein synthesis, emerging evidence indicates that it also exerts non-transcriptional effects. Specifically, priming regulates post-translational modifications of NLRP3, such as phosphorylation and ubiquitination, which are critical for controlling its activation. In its inactive state, ADP-bound NLRP3 may exist as an oligomer or a monomer. Monomeric NLRP3 localises to cellular membranes that act as scaffolds for assembly of an oligomeric double-ring structure composed of five to eight dimer pairs whose interlocking LRR domains form a back-to-back circular cage 60.
Anti-Inflammatory miRNAsMicroRNAs (miRNAs) are short non-coding RNA molecules of approximately 18–25 nucleotides that are transcribed by RNA polymerase II or III from intergenic or intragenic loci. Following initial processing by the RNase III enzyme Drosha in the nucleus, the precursor miRNA (pre-miRNA) is exported to the cytoplasm, where the endoribonuclease Dicer cleaves the hairpin to generate a miRNA duplex. One strand of this duplex is incorporated into the RNA-induced silencing complex (RISC), which modulates gene expression predominantly by promoting mRNA degradation or inhibiting translation. In certain contexts, however, miRNAs can stabilise target transcripts and even enhance their transcription or translation. Besides messenger RNAs, miRNAs have been reported to interact with small nuclear, ribosomal, transfer and long non-coding RNAs, although the functional significance of these interactions remains unclear. Because they are key regulators of haematopoiesis, immune-cell differentiation, immune responses, inflammation and autoimmunity, miRNAs offer a promising therapeutic avenue. Notably, miR-146 and miR-155 influence activation of the host defence system, thereby modulating immune regulation and inflammatory sequelae. Through miRNAs, both positive and negative regulatory events can affect the initiation, propagation and resolution phases of inflammation 61. Positive feedback initiates a series of molecular processes that limit microbial invasion and promote efficient tissue repair, whereas negative feedback, triggered only during severe inflammation, is essential to maintain tissue homeostasis and to prevent detrimental end-stage responses 62.
LimitationsCurrent pharmacological strategies for managing inflammation include corticosteroids, disease-modifying antirheumatic drugs (DMARDs), biologics, and non-steroidal anti-inflammatory drugs (NSAIDs). Although these agents are effective, they present several significant limitations:
Safety concernsBiologics: Agents such as tumour necrosis factor-α (TNF-α) antagonists (e.g., infliximab, adalimumab, and etanercept) have revolutionised the treatment of rheumatoid arthritis and inflammatory bowel disease. However, as noted by Hindryckx et al., their use is associated with an increased risk of infections, including the reactivation of latent tuberculosis, which is particularly problematic in immunocompromised individuals 63.
Traditional anti-inflammatory drugs: Prolonged NSAID therapy, while useful for short-term symptom control, is linked to gastrointestinal complications (e.g., ulcers, bleeding) and an elevated cardiovascular risk. Long-term corticosteroid administration, despite potent anti-inflammatory effects, may induce immunosuppression, osteoporosis, and metabolic disturbances.
Adverse effects with long-term use: Low-dose methotrexate (LD-MTX), a mainstay in rheumatoid arthritis, has been associated with interstitial lung disease, hepatotoxicity, and a heightened susceptibility to infection—particularly in patients with type 2 diabetes. A clinical trial of canakinumab highlighted these hazards, reporting a 67 % infection rate over two years compared with 25 % in the placebo group 64.
Efficacy limitationsVariable response: Clinical responses to anti-inflammatory therapy are heterogeneous, especially when treatment is initiated after disease onset. Sustained remissions are uncommon, and some patients develop neutralising antibodies against biologics, resulting in primary non-response or secondary loss of efficacy 65.
Disease-specific challenges: In atherosclerosis, anti-inflammatory approaches must surpass the benefits conferred by statins; moreover, inhibition of monocytosis may impair infarct healing and post-myocardial infarction remodelling.
Redundancy and compensation: Given the extensive network of cytokines and mediators, targeting a single pathway (e.g., TNF-α) may be insufficient. Compensatory mechanisms can exacerbate disease, as illustrated by murine studies in which TRAF6 or NF-κB blockade worsened atherosclerosis 66.
Future directionsPersonalised medicine aims to tailor therapy according to individual genetic and biomarker profiles to enhance efficacy and minimise adverse effects. For example, patients who are positive for anti-citrullinated protein antibodies (ACPA) or have elevated C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) often exhibit a more aggressive course of rheumatoid arthritis and may benefit from early, intensive interventions such as TNF inhibitors 67,68,69,70,71,72,73.
Comments (0)