
Remember me




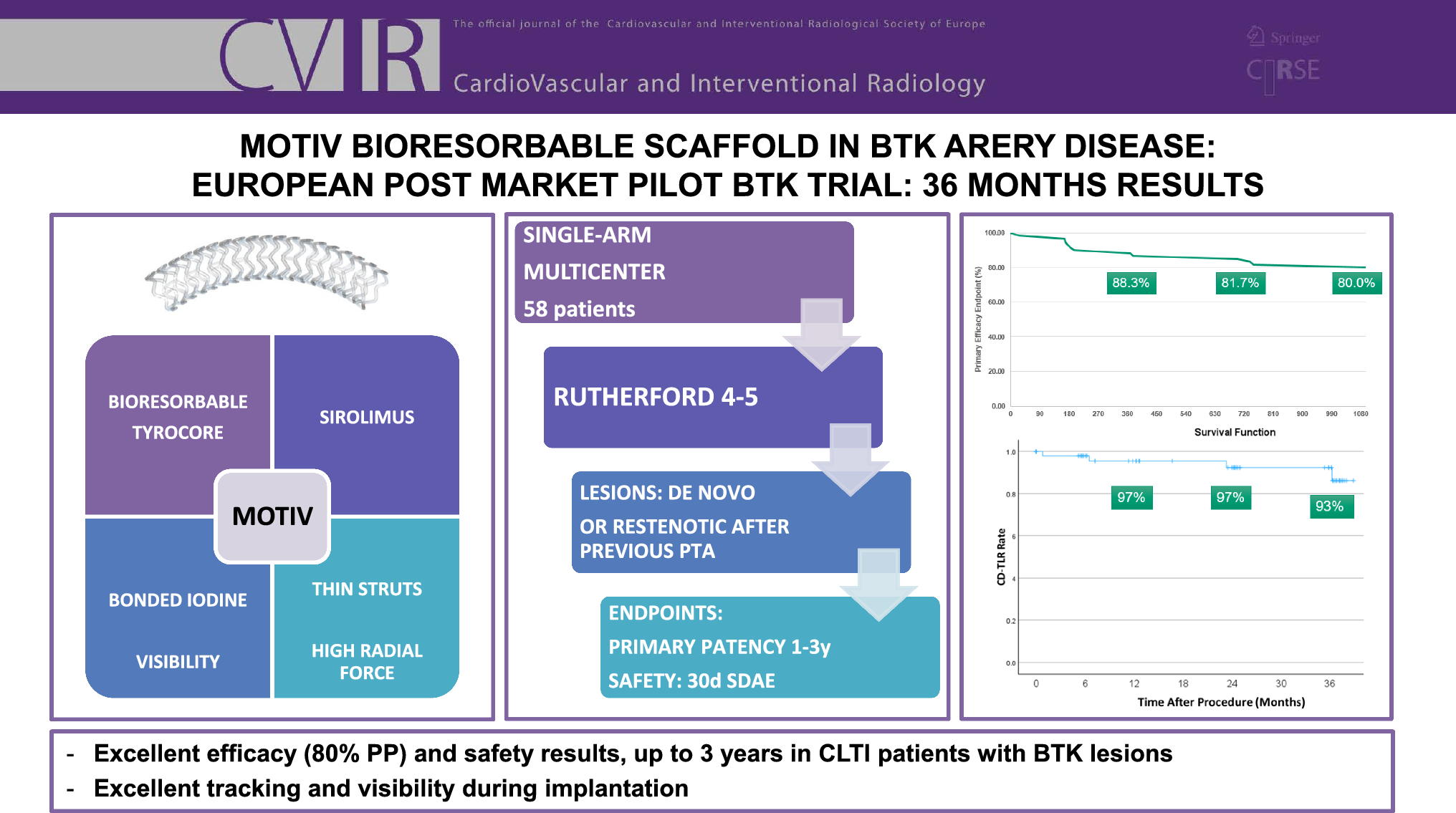
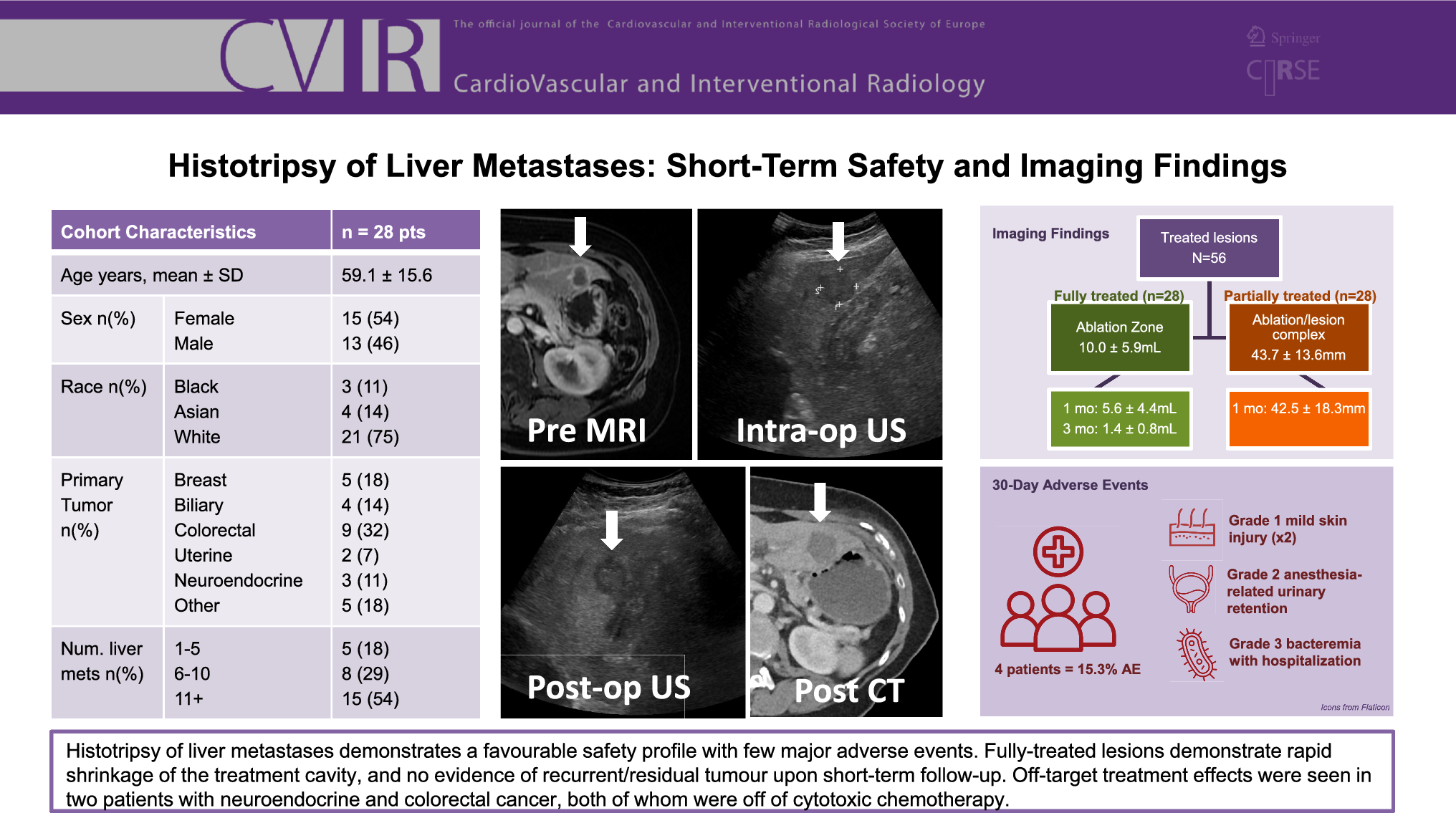
The MOTIV BTK pilot trial is a prospective, single-arm, multi-center, post-market study, designed to evaluate the safety and effectiveness of the MOTIV Peripheral Vascular Bioresorbable scaffold. The MOTIV scaffold is a combination device composed of Tyrocore, a bioresorbable material, coated with the antiproliferative drug Sirolimus at a concentration of 1.97 µg/mm2. Tyrocore is composed of tyrosine analogs (desaminotyrosine) and biocompatible hydroxy esters. It features an iodinated diphenol and a low-molecular-weight polylactic acid diol oligomer in an 8:1 ratio. The phenyl ring of the iodinated diphenol provides a robust molecular framework, contributing to Tyrocore’s exceptional tensile strength. Its ability to maintain flexibility while preserving strength stems from its distinctive composition and high molecular weight. The covalently bonded iodine enhances radiopacity, ensuring visibility under fluoroscopy. The strut thicknesses are 95 μm, 105 μm, and 115 μm for diameters of 2.5 mm, 3.0 mm, and 3.5 mm, respectively, and all devices are available in lengths up to 60 mm. The diameters from 2.5 to 3.0 mm may be safely post-dilated 0.75 mm beyond their nominal diameter, while 3.5 mm scaffolds can be expanded to 0.5 mm beyond their nominal diameter. After implantation, the MOTIV scaffold provides mechanical support for six months, as demonstrated by REVA Medical in a series of preclinical evaluations, and then gradually resorbs over four years. It is 6 French compatible and mounted on a rapid exchange balloon.

The MOTIV pilot study is registered at ClinicalTrials.gov under NCT03987061.
The trial planned to enroll 58 patients across nine clinical investigation centers. The name of the PI of each participating center can be found in Table 1. A sample size of 50 evaluable patients was chosen to provide an adequate estimate of “true ” rate of treatment success at 12 months, with an additional eight patients being added to offset for expected loss to follow-up and early study termination.
Table 1 PI and participating centerInclusion and exclusion criteria can be found in Table 2. In summary, patients with Rutherford 4 and 5 and BTK lesions could be enrolled. Two sorts of lesions were included in the study: de novo lesions requiring a scaffold (dissection or recoil) or restenotic lesions after previous PTA, suitable for endovascular therapy. Target vessel diameter estimated visually had to be ≥ 2.5 mm and ≤ 3.5 mm. The total lesion length that required scaffolding had to be < 100 mm. Longer lesions could be treated with angioplasty, as long as the total amount of scaffolding did not exceed 100 mm. In cases where total lesions coverage required two or more scaffolds, the MOTIV devices were placed in an edge-to-edge fashion, without overlap, under direct angiographic visualization. Inflow lesions could be managed at the operator’s discretion.
Table 2 In- and exclusion criteriaProcedureVascular access was obtained per the investigators’ standard clinical practice. Lesion crossing with the guidewire into the distal vessel was required prior to lesion preparation. All patients received 5,000 units of heparin at the start of the procedure.
Pre-dilatation was performed with a non- or semi-compliant balloon with a minimum reference to the artery ratio of 1:1 and with a maximum diameter of 3.5 mm under angiographic guidance.
Post-scaffold balloon dilatation was mandatory in all cases using a non- or semi-compliant balloon matching the reference diameter at ≥ 12 atm.
Operators were not blinded to treatment in follow-up assessments. All patients underwent clinical and duplex ultrasound follow-up at 1, 6, 24, and 36 months, with core lab adjudication. Data collected included patient demographics, medications, physical examination findings, ankle-brachial index (ABI), Rutherford score, and color flow Doppler ultrasound (pre-, intra-, and post-procedure measurements). Angiography, CT angiography (CT Angio), and MR angiography (MR Angio) were performed at the investigator’s discretion. Concomitant medication during hospital stays and follow-up was recommended as Clopidogrel 75 mg/daily for at least 3 to 4 months and lifelong aspirin: 75 to 300 mg daily.
Primary and secondary outcome measures were defined as:
Primary outcome measures:
1.Efficacy outcome measure: Primary patency rate (PPR) at 12 months, with PPR defined as no evidence of at least 50% restenosis or reocclusion within the originally treated lesion based on color flow duplex ultrasound (CFDU) measuring a peak systolic flow velocity ratio < 2.5 as determined by an independent Core Lab.
2.Safety outcome measure was defined as serious device-related adverse events within 30 days post-procedure.
Secondary outcome measures:
1.Technical success, defined as successful MOTIV implantation with a post-procedure residual angiographic stenosis < 30%
2.PPR rate a 1-, 6-, 24-, and 36-month follow-up as defined in the efficacy outcome measure above.
3.Clinical driven target lesion revascularization (CD-TLR) as a repeat intervention to maintain or re-establish patency within the region of the treated arterial vessel plus 5 mm proximal and/or distal to the treated lesion
4.Limb salvage rate, defined as the absence of major amputation.
5.Clinical success at follow-up was defined as an improvement of Rutherford classification at follow-up of at least one class compared to the pre-procedure Rutherford classification.
Comments (0)