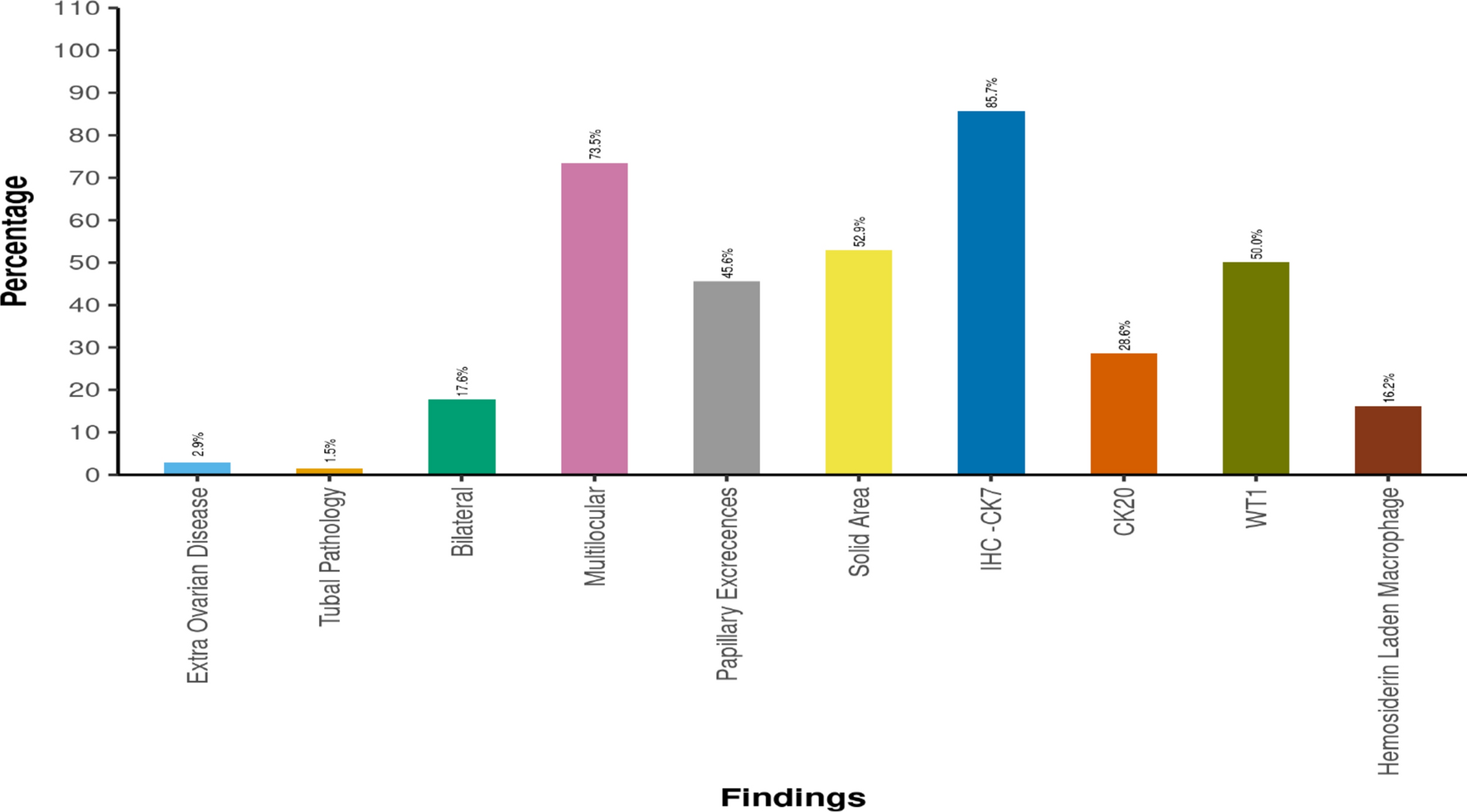
The source of endometrial cancer tissue specimens
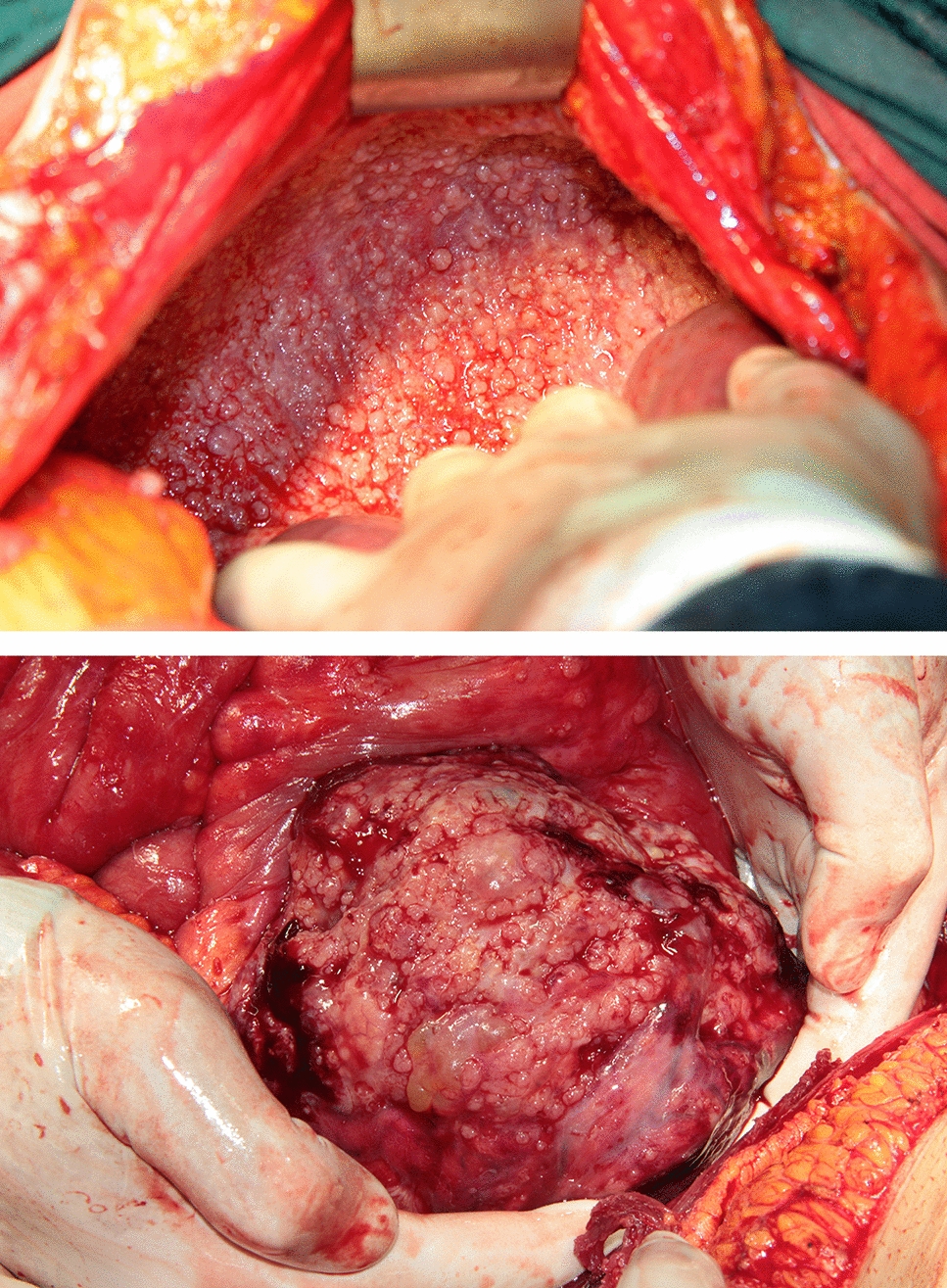
Endometrial cancer tissue specimens were acquired from ten fresh endometrial cancer tissue samples of patients undergoing comprehensive staging surgery for endometrial malignant tumors at the Gynecology Department of the Third Affiliated Hospital of Sun Yat-sen University between September 2023 and January 2024. None of the patients received neoadjuvant therapy (including chemotherapy and radiotherapy) prior to surgery. Clinical data and histopathological characteristics were retrieved from medical records and routine pathology reports. The study was approved and supervised by the Third Affiliated Hospital of Sun Yat-sen University. Informed consent was obtained from all endometrial cancer patients. Specimens were transported on ice from the hospital to the laboratory of Tsinghua–Berkeley Shenzhen Institute (TBSI) on the day of collection.
Preparation of Endometrial Cancer Organoid SamplesSample Preprocessing
Freshly obtained endometrial cancer specimens, procured immediately post-surgery in the operating room, were preserved and conveyed on ice. These samples were then subjected to a 20-min incubation period in pre-heated (37 °C) DMEM/F12 washing medium enriched with l-glutamine, penicillin–streptomycin, and HEPES.
Utilizing forceps, the tissue sample was carefully transferred from the preservative solution to a sterile culture dish. Subsequently, the endometrial cancer tissue underwent thorough cleansing with chilled PBS (containing 1% bicarbonate) five to ten times. Placing the tissue specimen into a sterile dish, a minimal volume of DMEM/F12 was added to ensure the tissue remained hydrated. A surgical scalpel was employed to finely mince the tissue, ensuring the removal of any adipose, normal, vascular, and damaged tissue components from the sample. The minced tissue was reduced to pieces measuring 0.5–1 mm in size on an ice-cold platform.
Three aliquots of tissue were set aside within separate 1.5-ml centrifuge tubes: one designated for DNA extraction; another, prepped with RNAse inhibitors, was reserved for RNA extraction, both labeled and transferred to − 80 °C for cryopreservation. The final aliquot received 1 ml of 0.4% paraformaldehyde (PFA), which was appropriately labeled, and stored at 4 °C for further use.
Organizational Digestion
The dissected tissue is collected in a 15-ml centrifuge tube using cold PBS (containing 1% bispecific antibody), centrifuged at 1200 rpm for 5 min and discarded the supernatant. 3–5 ml of digestion solution I is added to the pellet and digested within a 37 °C constant temperature shaker for 40–60 min. Throughout digestion, the tissue’s digestion status is inspected every 15–20 min, promptly terminating digestion when the tissue appeared dispersed. Following digestion, a 1-ml pipette tip is used to blow and mix the digestion solution 10–20 times, centrifuged at 1200 rpm for 5 min and discarded the supernatant. 2–3 ml of digestion solution II was added and continued digestion in a 37 °C constant temperature shaker for approximately 5–10 min. During this period, the centrifuge tube is inverted several times every 3 min. The tissue’s digestion under a microscope is observed, gently blew and beat the digestive fluid around 10–20 times. Digestion is terminated when the tissue had been reduced to smaller cell clumps. The volume of PBS (containing 1% bispecific antibody) is added twice to halt digestion, blew and mixed around 10–20 times.
Filter and Collect Cells
The filtrate is filtered and collected using a 100-μm filter. The filter is rinsed with 2–5 mL of PBS (containing 1% double antibody) post-filtration, centrifuged at 1200 rpm for 5 min after collecting the filtrate and discarded the supernatant. 3–5 mL of red blood cell lysis buffer is introduced to the cell pellet, pipetted vigorously to ensure thorough mixing, and incubated at room temperature for 3–5 min. The lysis process is terminated by adding an equal volume of PBS (containing 1% bispecific antibody), centrifuged at 1200 rpm for 3 min and discarded the supernatant.
Preparation of Cell Suspension
The cell pellet in 1 mL of advanced DMEM/F12 medium is resuspended and proceeded with cell counting. The suspension is centrifuged at 1200 rpm for 3 min and gently aspirated the supernatant without disturbing the cell pellet.
Cell Inoculation and Culture
The matrix gel and cell suspension were thoroughly mixed at a ratio of 1.2:1 (resulting in a final matrix gel concentration of 5.5 mg/ml) while maintaining a cell density of 500–1000 cells/µl. Using a pipette gun, 20–30 µl of the gel-cell mixture was aspirated and inoculated into the center of each well in a 24-well plate, leading to the formation of a distinct dome structure within each well. Following inoculation, the plate was placed in an incubator set at 37 °C and allowed to rest for 5 min. To facilitate even gel solidification, the plate was gently shaken without disturbing the droplets, after which it was inverted and left undisturbed for an additional 25 min until the gel droplets were fully solidified. Subsequently, 800–1000 µl of organoid expansion medium was added to each well, and the cultures were maintained in the incubator. Throughout the cultivation period, the progress was closely monitored, with media changes carried out every 2–3 days as necessary.
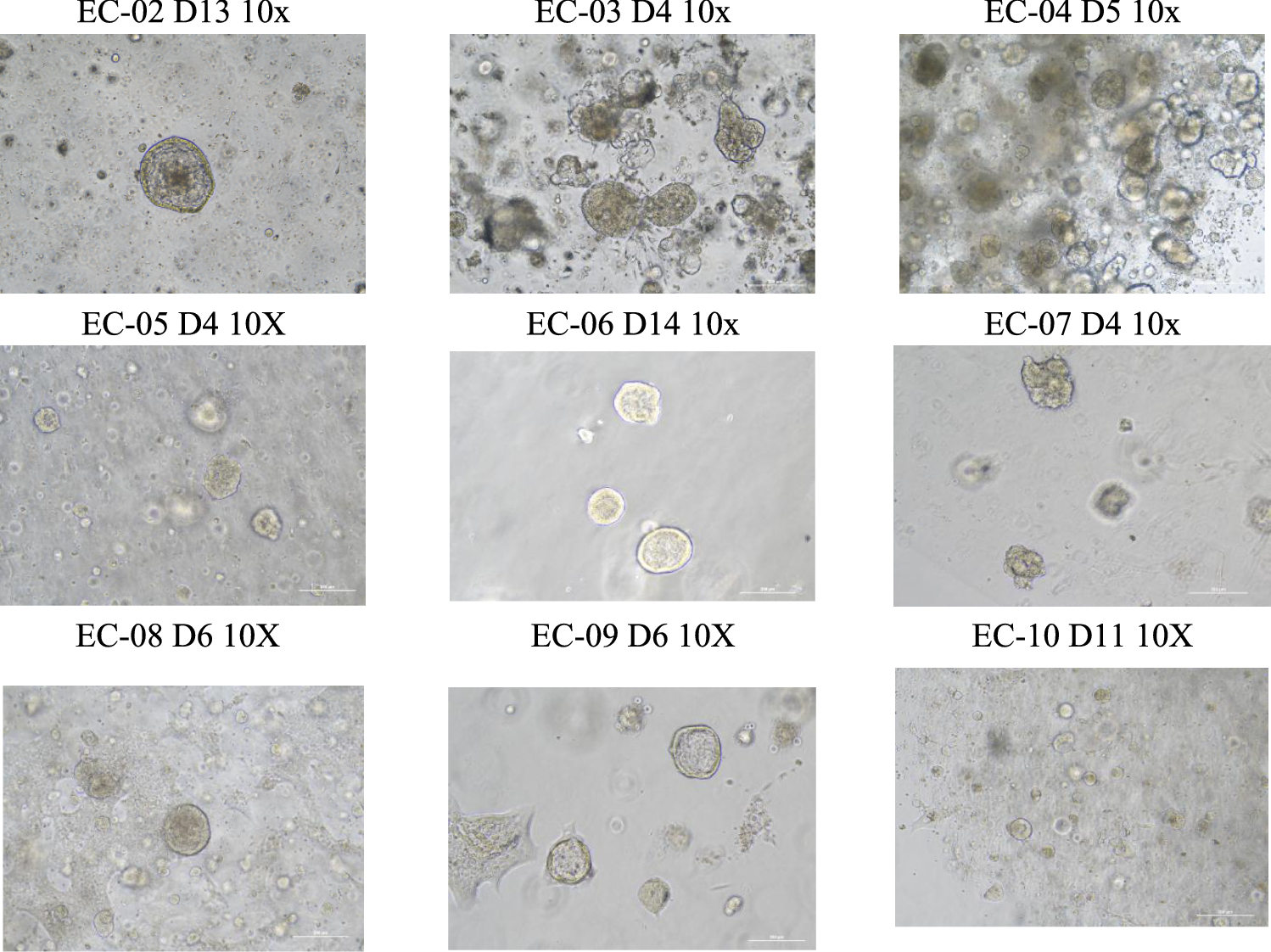
Endometrial Cancer Organoid Subculture
Passaging should be promptly performed when organoids reach a diameter of 100-200 μm, as nutrient deprivation and subsequent acidification (evidenced by rapid yellowing of the culture medium) may induce necrotic core formation within the aggregates. Gently aspirate the culture medium from the Petri dish without harming the Matrigel dome. To each well, add 1 ml of TrypleE, disrupt the matrix gel using a 1 ml pipette tip, and incubate at room temperature for 3–5 min to digest the organoids into cell spheres comprising 2–3 cells (diameter less than 20 μm). Transfer the organoids from the well plate to a 15-ml centrifuge tube, subsequently adding 2–3 times the volume of DMEM/F12 to halt digestion. Simultaneously, take 1 ml of DMEM/F12 to rinse the corresponding well of the Petri dish and transfer it to a 15-ml centrifuge tube as well and centrifuge at 1200 rpm for 5 min, discarding the supernatant; use organoid culture medium for cell resuspension. If the organoid’s condition is favorable, passage is executed at a 1:3 ratio; if the organoid’s state is less than optimal, passage should occur at a 1:1 ratio.
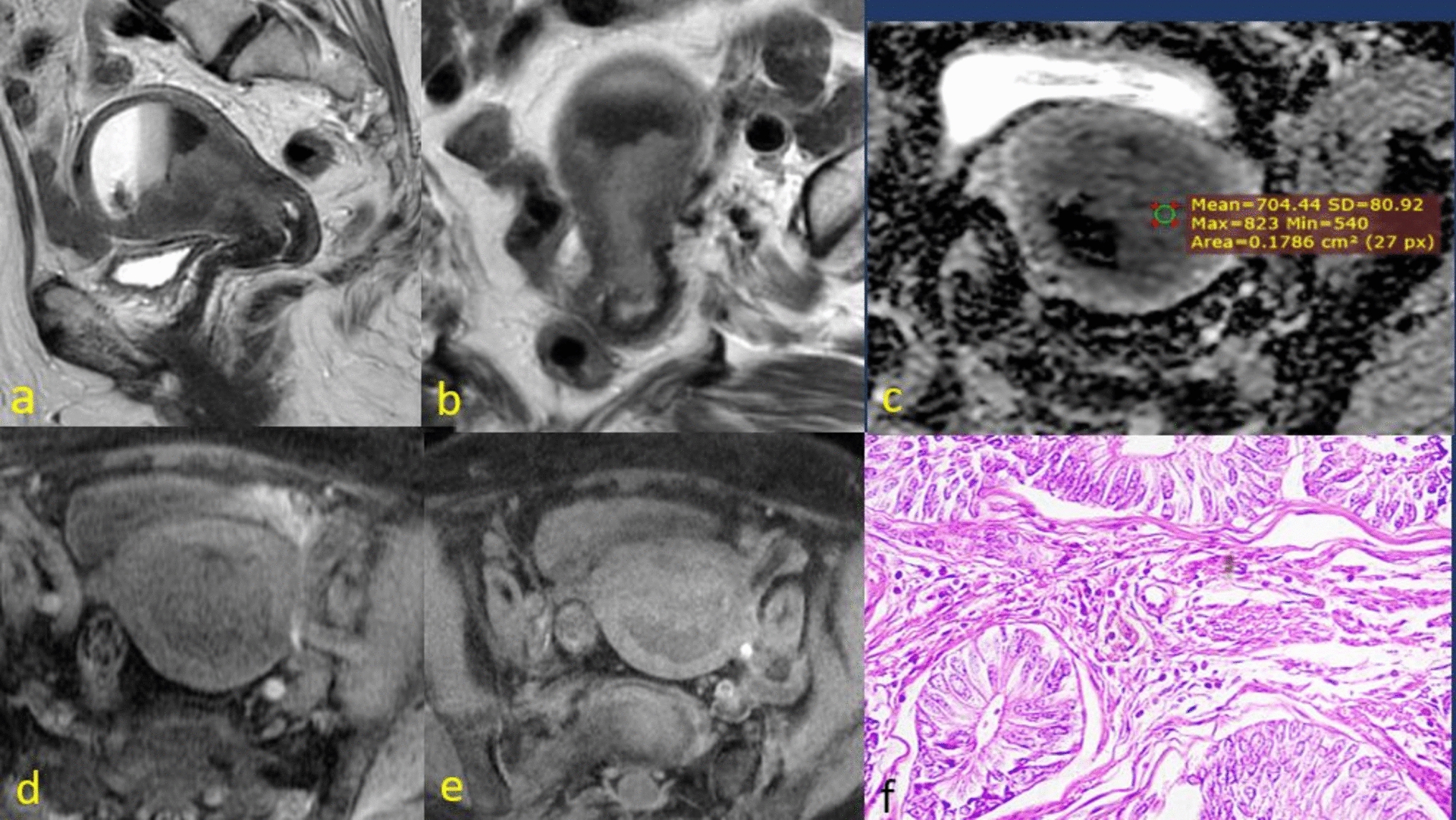
Identification of Endometrial Cancer Lesion Tissue and OrganoidsHematoxylin Eosin Staining (HE)
Collect endometrial cancer organoid cells and let them stand at 4 °C for 10 min. Centrifuge and discard most of the supernatant. Weigh 0.2–0.4 g of agarose and add it into a small reagent bottle. Add 10 mL of phosphate buffered saline (PBS) and melt the agarose in high power in the microwave oven. Take 200 μl of the melted agarose and add it to a 1.5-ml EP tube. Form a deep-rounded bottom groove on the surface of the agarose. Aspirate the organoid sediments and place them at the bottom of the depression, embedding them in the middle position. Remove the agarose organoid embedding block after it has solidified and dehydrate it in gradient ethanol. Preheat the drying oven about 4 h in advance and melt the wax at 65 °C. Transfer the dehydrated agarose block to an embedding box and move the box to xylene for 5 min twice for clarification. Immediately transfer to the melted wax container, keep it immersed in wax at 60 °C for 2 h, then change to a new wax container for another 2 h of immersion. Embed on ice, and wait until the wax block is completely solidified before removing the mold. The lesion tissues are fixed in 4% paraformaldehyde solution for 48 h, and then placed in the embedding frame with molten wax for embedding. Wax sections are sequentially treated with xylene, absolute ethanol, 95% alcohol, etc., for dewaxing, distilled water washing. Place the sections in Harris’ hematoxylin for 3–8 min, wash with tap water, differentiate for a few seconds with 1% hydrochloric acid alcohol, wash with tap water, bluing with 0.6% ammonia water, wash with running water. Then, stain the sections with eosin for 1–3 min, and sequentially put the sections into 95% alcohol, absolute ethanol, and xylene for dehydration to transparency. Remove the sections from xylene and slightly dry and seal with neutral gum. Examine under the microscope and image acquisition analysis.
Immunohistochemistry (IHC)
Paraffin embedding and sectioning steps are the same as before. When preparing for dewaxing, place the sections in an oven at 60 °C for over 4 h. Immerse the sections in xylene, followed by alcohol baths. Finally, soak the sections in double-distilled water for 2 min. Choose an endogenous peroxidase blocker and apply for 20 min. Soak three times in double-distilled water, each for 5 min. Prepare 400–600 mL of citric acid antigen retrieval solution (pH = 6.0, 10 nM), immerse the slides, and then microwave on high for 10 min. After heating, replenish with double-distilled water, then microwave on low for 40 min, topping up every 10 min. Post-retrieval, cool to room temperature naturally. Ultra-clean water wash followed by PBS three times, each for 3 min. Incubate with primary antibody overnight at 4 °C, then warm to 37 °C the next day, followed by PBS wash for 5 min. Add enhancer solution, incubate at room temperature for 20 min, then PBS wash three times, each for 3 min. Add secondary antibody, incubate at room temperature for 20 min, then PBS wash three times, each for 3 min. Apply DAB chromogen, incubate at room temperature for 5–10 min, then gently rinse off with tap water. Stain with hematoxylin, wash under running water for 5 min. Sequentially immerse in gradient alcohols, then process with xylene, each step for 20 min. Seal with neutral resin and air-dry naturally.


Comments (0)