
Remember me
You have full access to this article via your institution.
In a pivotal study, Wang and colleagues reveal how early life high fructose exposure disrupts neurodevelopment. The publication uncovers the GLUT5-HK2 axis by which excessive fructose exposure during postnatal development impairs physiological microglial pruning.
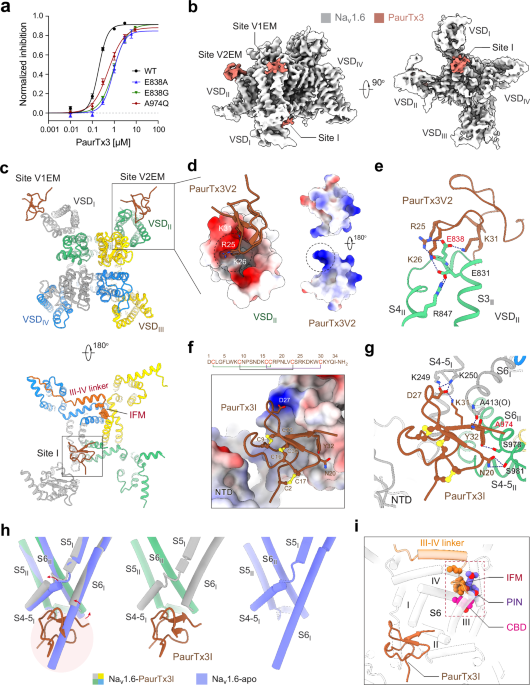
Microglia, the embryonically-derived tissue-resident macrophages of the central nervous system (CNS) parenchyma, have long been implicated in neurodevelopment as shapers of neuronal circuitry.1 They are established early in the embryonic development of the CNS and constantly renew themselves without replacement by bone marrow-derived cells in the steady-state CNS. They are known not only to shape neuronal circuits in development and adulthood, but also to provide immune surveillance, supply trophic support for neurogenesis and the blood–brain barrier, and directly regulate neuronal activity.2,3
Slc2a5, the gene encoding the fructose-selective transporter GLUT5, is specifically expressed in microglia in the brain and belongs to the homeostatic transcriptomic core signature of these cells. Even though the expression of Slc2a5 in microglia and even its postnatal upregulation had been described previously,4 a function for this transporter in microglia had to date not been elucidated.
Research of the last decades suggests that excessive fructose or sugar consumption during pregnancy or childhood, largely derived from food additives in the standard American diet, correlates with negative mental health outcomes. However, a causative relation or mechanistic insight had not been established previously.5
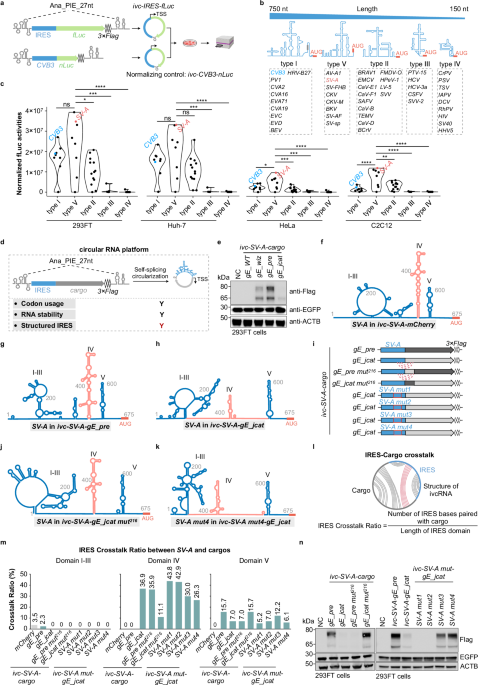
To address whether fructose has an effect on CNS development in mice, Wang and colleagues placed pregnant wild-type or Slc2a5-deficient dams on isocaloric diets containing either 15% or 0% fructose (Fig. 1).6 Since the prefrontal cortex (PFC) prominently develops throughout prenatal and neonatal stages and disturbances in this critical phase can lead to impaired social behavior and cognition,7 the authors histologically assessed the PFC of 1-week-old offspring which revealed an accumulation of uncleared apoptotic TUNEL+ cells in fructose-receiving mice. Since microglia are the singular cell type expressing Slc2a5 in the homeostatic CNS with a well-known role in the structural and functional adaptations of the PFC during this period,8 they next quantified microglial phagocytosis and found a reduction of postsynaptic density protein 95 (PSD95) inside microglial lysosomes and increased PSD95 density in the PFC.
Fig. 1: GLUT5 facilitates fructose uptake into microglia, triggering a metabolic shift toward fructolysis and upregulation of HK2.
Following fructose breakdown, physiological microglial phagocytosis is disrupted, leading to the accumulation of dead neurons and unreinforced synapses in the PFC during development, ultimately resulting in cognitive deficits in juvenile mice.
This prompted the authors to investigate microglial phagocytic capacity in response to fructose exposure. The following in vitro testing of cultured primary microglia from neonatal mice or human induced pluripotent stem cell (iPSC)-derived microglia with or without fructose revealed upregulation of GLUT5 and confirmed decreased phagocytosis of apoptotic neurons and synaptosomes in response to fructose. This decreased phagocytic capacity was reversed in microglia derived from mice lacking Slc2a5. Taken together, fructose exposure led to dampened microglial phagocytic activity both in vivo and in vitro.
As a simple carbohydrate, fructose is metabolized within cells to generate energy, but in excess, it may alter cellular metabolism to accommodate the overabundance. To assess fructose metabolism in the developing brain, the authors used [2-13C]-fructose carbon nuclear magnetic resonance after injecting [2-13C]-fructose into neonates born to dams on high fructose or control diet. The metabolic fate of the labeled fructose strikingly differed between the groups, with the fructose-exposed group preferentially breaking down fructose into lactic acid and glutamic acid. Furthermore, isotope-labeled fructolysis and tricarboxylic acid cycle intermediates were found in greater abundance in the fructose-exposed group, while ATP concentration was decreased, indicating a fructose-mediated metabolic shift. These metabolic adaptations were absent in brains from Slc2a5–/– mice.
A recent study demonstrated a role for hexokinase 2 (HK2) in negatively regulating microglial phagocytosis by shifting metabolism toward fructolysis and glycolysis and decreasing intracellular ATP.9 Wang and colleagues found that fructose exposure increased the expression of HK2 in primary microglia and inhibiting HK2 rescued the decrease in phagocytic activity elicited by fructose exposure. In combination, these data demonstrate that overabundance of fructose leads to a metabolic shift and increased HK2 activity in microglia which result in a less phagocytic microglial state.
In a subsequent step, the authors investigated the relevance of these fructose-mediated changes in the PFC for the neurological performance of adolescent mice. Therefore, wild-type or Slc2a5-deficient mice born to and nursed by dams on high fructose or control diet were behaviorally assessed after weaning (~4 weeks of age). Wild-type mice exposed to fructose showed decreased novel object preference, suggesting impaired cognition, and inability to override fear learning in a fear extinction paradigm, indicating cognitive inflexibility. These behavioral abnormalities were absent in mice lacking Slc2a5.
Notably, up until this point, the globally Slc2a5-deficient mice deployed as a control condition for the effects of fructose on microglia lacked cell type specificity, limiting the significance of the experiments. Hence, in a final step, the authors replicated their key histological, metabolic and behavioral findings in a myeloid-specific Csf1rCre;Slc2a5fl/fl mouse line. Together, these findings reveal that microglial function exhibits a metabolic sensitivity to fructose excess, which, during neurodevelopment, leads to impaired cognition and memory extinction in juvenile mice.
Neurodevelopment is a finely tuned process in which structural organization and cellular division and movement occur along predefined stages. Disturbances of this delicate procedure can have a long-lasting impact on an organism. Wang and colleagues for the first time provide mechanistic insight into how overabundance of simple sugars, specifically fructose, diverts neurodevelopment by shifting microglial metabolism and function, thereby inhibiting the necessary clean-up activity that they provide to immature neuronal networks. Strengths of their study approach include the use of clinically-relevant dietary concentrations of fructose in their in vivo experiments, the provided mechanistic insight pertaining to the altered metabolism of microglia and their validation of the dependence of the effects on Slc2a5 expression in myeloid cells. Excitingly, the authors describe a relevant role for a core gene of homeostatic microglia during fructose overexposure, even though a potential function under homeostatic conditions is still left to be deciphered. Additionally, this prompts the question of why microglia acutely regulate brain fructose levels and whether this process plays a significant role in brain function.
While the authors show that human iPSC-derived microglia respond to fructose similarly to primary mouse microglia, caution is warranted in directly translating these findings, as the clinical evidence remains limited.
Future studies could look into the long-term absence of Slc2a5 in microglia by utilizing microglia-specific and inducible genetic tools10 to uncover a potential homeostatic function for this microglial core gene. Since metabolic shifts in microglia have been implicated in neurodegenerative disease models,11 studying the role of microglial metabolism and phagocytosis through the lens of GLUT5 expression and HK2 function could offer new insights into disease functions of the brain’s central immune sentinels.
Comments (0)