Cell culture
IPEC-J2 cells were grown in complete culture medium containing Dulbecco’s modified Eagle’s medium /nutrient mixture F12 (DMEM/F12, Cat NO. 11320033, Gibco), supplemented with 10% fetal bovine serum (FBS, Cat NO. A3160801, Gibco) and 1% penicillin/streptomycin. MA104 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Cell transfection
A total of 1 × 106 IPEC-J2 cells were seeded in a 6-well plate and cultured in an incubator at 37 °C with 5% CO2 for 24 h. For siRNA transfection, Lipofectamine™ RNAiMAX (Invitrogen, Cat NO. 13778075) was used. Specifically, 9 µL of RNAiMAX was diluted in 150 µL of Opti-MEM and thoroughly mixed by pipetting. Simultaneously, 3 µL of siRNA was diluted in 150 µL of Opti-MEM and mixed well. The diluted siRNA solution was then added to RNAiMAX mixture and incubated at room temperature for 5 min. Subsequently, cells were rinsed once with Opti-MEM, and 250 µL of the siRNA and RNAiMAX mixture was added to cells, followed by the addition of 750 µL of complete culture medium. The cells were cultured in an incubator at 37 °C with 5% CO2 for 24–48 h before samples collection. The siRNA sequences used were listed in Supplementary Table 1. For plasmid transfection, Lipofectamine 3000 (Invitrogen, Cat NO. L3000015) was employed. Specifically, 7 µL of lipofectamine 3000 was diluted in 125 µL of Opti-MEM and thoroughly mixed by pipetting. At the same time, 2.5 µg of plasmid and 5 µL P3000 were diluted in 125 µL of Opti-MEM. The plasmid solution was then added to lipofectamine 3000 mixture and incubated at room temperature for 15 min. Subsequently, the cells were rinsed once with Opti-MEM, and 250 µL of the plasmid and lipofectamine 3000 mixture was added, followed by the addition of 750 µL of complete culture medium. The Flag-tagged human Mettl3 plasmid and Flag-tagged porcine Ythdf2 plasmid were generous gifts from Dr. Chuan He (University of Chicago) and Dr. Xinxia Wang (Zhejiang University), respectively. The Ifi44l and Irf2 plasmids were generated by cloning the full-length open reading frames of porcine Ifi44l (XM_003127919.4) and Irf2 (XM_005671703.3) into pcDNA3.1 mammalian expression vector, which were purchased from Keygen Biotech. The full-length cDNA sequence of porcine Mettl3 genes was amplified by reverse transcription-polymerase chain reaction (RT-PCR) using forward primer: 5’- acctccatagaagatAATGTCGGACACGTGGAGC.
TCTA-3’ and reverse primer: 5’- GATCCATTTAAATTCgaattcTGGCCAT.
GGATGCTTAGGTTTC-3’. The PCR product was then ligated to the CMV-MCS-EF1-mcherry vector, which was previously digested with XbaI and EcoRI using CloneEZ Enzyme (Genscript, China) [15].
Infection and propagation of porcine rotavirus
The G9P [7] strain of Porcine rotavirus (PoRV) NJ2012 (NCBI accession number: MT874983-MT874993) and Nsp5 gene positive standard were kindly provided by Professor Li Bin from Zoonosis Prevention and Control Laboratory at Veterinary Research Institute of Jiangsu Academy of Agricultural Sciences. The PoRV, dissolved in DMEM/F12, was activated with Trypsin (10 µg/mL) at 37 °C for 30 min. The activated virus (multiple of infection = 0.1) was added to the IPEC-J2 cells that had reached 90% confluence and incubated at 37 °C with 5% CO2 for 1 h, with slight shaking every 20 min to ensure uniform virus adsorption. After adsorption, cells were maintained in complete culture medium containing 1 µg/mL Trypsin at 37 °C and 5% CO2 until over 85% cytopathic effect (CPE) was observed.
PoRV was propagated in MA104 cells using a similar infection method as employed for IPEC-J2 cells. After infection, the MA104 cells showing CPE underwent three cycles of freezing and thawing, alternating between − 80 °C and room temperature. Subsequently, the mixture was centrifuged at 5,000 rpm for 10 min to remove cell precipitation, and the supernatant was collected as the propagated virus.
Purification of PoRV
The purification of PoRV was performed using the sucrose density gradient centrifugation method. Initially, virus supernatant was centrifuged at 100,000 rpm for 2 h. The supernatant was discarded, and the pellet was resuspended in 100 µL of 1×PBS buffer and incubated overnight at 4℃. A 20% sucrose solution (15 mL) was added to a new 50 mL centrifuge tube. Subsequently, 15 mL of a 60% sucrose solution was slowly added to the bottom of the centrifuge tube using a long needle, creating an interface between the two sucrose concentrations. The virus solution was then slowly added above the 20% sucrose solution, forming another interface. The tube was centrifuged at 100,000 rpm for 2 h. After centrifugation, the visible band at the interface between the sucrose solutions was carefully aspirated. The aspirated band was diluted in 1×PBS buffer at a ratio of 1:5 and subjected to centrifugation at 100,000 rpm for 1 h. The supernatant was discarded, and the pellet was resuspended in 100 µL of 1×PBS buffer. The resuspended virus was stored at -20℃ for MeRIP-seq.
Assessment of PoRV titer
The PoRV titer was determined using the 50% tissue culture infective dose (TCID50) method [16]. The virus was dissolved in DMEM and serially diluted 10-fold, ranging from 10− 1 to 10− 10. Subsequently, the diluted virus was inoculated into MA104 cells in a 96-well plate, with two columns of uninfected cells serving as negative controls. After 24 h, CPE was observed, and the viral titer was calculated using the 50% cell culture infectious dose TCID50.
Determination of PoRV RNA copy number
The positive standard for PoRV Nsp5 gene was diluted 10-fold, and DNA concentration was assessed using a micro-spectrophotometer. Subsequently, a dilution series ranging from 10− 5 to 10− 12 was prepared based on standard concentration. Eight templates derived from this dilution series were utilized as positive standard templates for qRT-PCR. The linear relationship between cycle threshold (CT) value and copy number was analyzed to obtain the copy number standard curve. In the subsequent experiments, the PoRV Nsp5 gene copy number was calculated by inputting the CT value into the standard curve. The primer sequence for Nsp5 was provided in Supplementary Table 2.
RNA extraction and gene expression determination
Cells were lysed in 1 mL of RNA isolation reagent (Accurate Biotechnology, Cat NO. AG21102). Approximately 200 µL of trichloromethane solution was added to separate the organic and aqueous phases. Then, about 400 µL of supernatant was transferred into a new 1.5 mL enzyme-free centrifuge tube and mixed with an equal volume of pre-cooled isopropyl alcohol solution to precipitate RNA. The RNA precipitate was rinsed twice with 75% ethanol solution. After that, the RNA was dissolved in sterile, enzyme-free water. The integrity of RNA was verified by agarose gel electrophoresis, and the concentration and purity of total RNA were determined using a micro-spectrophotometer.
A total of 1 µg of RNA was reverse transcribed into cDNA using the PrimeScript @ RT kit (Accurate Biotechnology, Cat NO. AG11728). The qPCR reaction reagents consisted of the following: 5.0 µL of 2×ChamQ SYBR qPCR Master Mix (Accurate Biotechnology, Cat NO. AG11718), 0.2 µL of each of forward and reverse primers (10 µmol/L), 2.0 µL of cDNA, 2.6 µL of ddH2O. The qPCR program was as follows: pre-denaturation at 95℃ for 30 s, followed by 40 cycles of 95℃ for 10 s and 60℃ for 30 s. GAPDH was used as the internal reference gene. The relative expression levels of the target gene were calculated using the 2−ΔΔct method. The primer sequences were listed in Supplementary Table 2.
MeRIP-seq
The experiment was carried out according to the procedure previously reported [17, 18], with three independent biological replicates per experimental group to ensure reproducibility. mRNA was isolated from total RNA using the Dynabeads™ mRNA DIRECT™ kit (Ambion, Cat NO. 61012). The purified mRNA was then fragmented by 30 cycles of ultrasound, with each cycle comprising 30 s on and 30 s off. The concentration of fragmented mRNA was determined using a Qubit™ RNA HS assay kit (Invitrogen, Cat NO. Q32852). Next, 500 ng of mRNA was taken as the input control for sequencing. A total of 950 ng of fragmented mRNA was incubated with 1 µL of m6A antibody (NEB, Cat. E1610S) at 4 °C for 1 h. After incubation, the mixture was washed twice in reaction buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40), twice in low salt buffer (50 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40), and twice in high salt buffer (500 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40). Bound mRNA was eluted using 100 µL of elution buffer (QIAGEN, Cat. 1015762).
Library preparation was conducted using the SMARTer Stranded Total RNA-Seq Kit version 2-Pico Input Mammalian (Takara) per the manufacturer’s instructions. Briefly, the immunoprecipitated mRNA fragments and an equal amount of input control were subjected to cDNA synthesis using the kit components SMART Pico Oligos Mix v2, 5X First-Strand Buffer, SMART TSO Mix v2, and SMARTScribe Reverse Transcriptase. The cDNA was then ligated with Illumina adapters and indexes, followed by PCR amplification. The libraries were then purified using AMPure beads to remove excess primers and nucleotides. Ribosomal cDNA removal was performed using the ZapR v2 and R-Probes v2 reagents provided in the kit. The library fragments were subjected to a second round of PCR amplification to generate sufficient quantity for sequencing. Before sequencing on an Illumina HiSeq4000, the libraries were analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies).
For MeRIP-seq analysis, to validate the specificity of the identified peaks, a negative control (uninfected cells) is included in MeRIP-seq experiment. The differential m6A peaks from single-factor comparisons between Mock and PoRV groups were analyzed using the R package exomePeak2 (version 1.8.1). A Poisson generalized linear model (GLM) was employed as a quantitative method to identify m6A peaks within individual groups and m6A peaks of single-factor comparative differences between groups. To minimize false positives, the identified m6A peaks from each biological replicate were deemed significant based on the following thresholds: peak width < 1500 bp, log2 fold change (log2FC) > 1, false discovery rate (FDR) < 0.05, Input RPM > 0.1, and IP RPM > 0.1. Differential m6A peaks between groups were considered significant under these criteria: peak width < 1500 bp,|diff.log2FC| > 1, FDR < 0.05, Input Control RPM > 0.1, IP Control RPM > 0.1, Input Treated RPM > 0.1, and IP Treated RPM > 0.1.
Measurement of M6A content
The total RNA concentration was adjusted to 100 ng/µL. M6A content was measured using the EpiQuikTM m6A RNA Methylation Quantification Kit (Epigentek, Cat. P-9005). First, 80 µL of binding solution was added to each well. Then, 2 µL of negative control reagent, 2 µL of positive control reagent, and 2 µL of the sample were added to the negative control wells, positive control wells, and sample wells, respectively. The plate was incubated at 37 °C for 90 min. After incubation, the liquid was discarded, and each well was washed three times with 150 µL of wash buffer. M6A RNA was captured by incubation with 50 µL of diluted capture antibody at room temperature for 60 min, followed by incubation with 50 µL of diluted detection antibody at room temperature for 30 min, and finally incubation with 50 µL of diluted enhancer solution at room temperature for 30 min. After each incubation step, the liquid was discarded, and each well was washed with 150 µL of wash buffer. Subsequently, 100 µL of developer solution was added to each well and incubated at room temperature away from light for 7 min. Finally, when the color in the positive control turned moderately blue, 100 µL of stop solution was added to each well to terminate the enzyme-catalyzed reaction. The absorbance at 450 nm was measured.
Extraction of nuclear protein and cytoplasmic protein
Nuclear protein and cytoplasmic protein were extracted according to the manufacturer’s guidelines (Beyotime, P0027).
Total protein extraction and western blot
A total of 200 µL of RIPA cell lysis buffer (Beyotime, Cat NO. P0013B) and phenylmethanesulfonyl fluoride (PMSF) were added to each well of a 6-well plate. After 30 min, the lysed cells were transferred to 1.5 mL enzyme-free centrifuge tubes and centrifuged at 4℃ at 12,000 x g for 10 min. The supernatant was transferred to new 1.5 mL enzyme-free centrifuge tubes, and the protein concentration was determined using a BCA protein assay kit (Beyotime, Cat NO. P0010). After standardizing protein concentration, 5× loading buffer (Beyotime, Cat NO. P0015) was added, and the protein was denatured at 95℃ for 10 min using a metal bath. The content of target protein was determined by Western Blot. Denatured proteins were separated by SDS-PAGE gel electrophoresis. After electrophoresis, proteins were transferred from the gel to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked in a 5% skim milk solution at room temperature on a shaker for 2 h. The membrane was washed 3–5 times with Tris-buffer solution containing Tween-20 (TBST) on a shaker for 5 min each. The membrane was then incubated with primary antibody overnight at 4 °C on a shaker. After washing the membrane again, secondary antibody was incubated at room temperature on a shaker for 90 min. Immunoblots were visualized using enhanced chemiluminescence. β-actin was used as the internal control. Antibody information was presented in Supplementary Table 3.
Immunofluorescence
The cells were fixed with 4% paraformaldehyde for 10 min at room temperature and then permeabilized with 0.3% Triton X-100 in PBS for 20 min. Protein blocking was performed using 3% bovine serum albumin for 60 min. The cells were incubated overnight at 4 °C with primary antibodies, followed by a 1-h incubation at room temperature with Alexa series fluorescently labeled secondary antibodies. After washing with PBST, nuclei were detected using DAPI. Images were captured using a LSM710 confocal laser scanning microscope (Carl Zeiss) and analyzed with ImageJ software.
Co-immunoprecipitation
293T cells were seeded into 12-well plates and cultured overnight in a 37 °C, 5% CO2 incubator. Upon reaching 70% confluency, cells were transfected with either METTL3 plasmid or empty vector control. After 24 h of transfection, all wells were infected with 0.1 MOI PoRV for 12 h. Cells were then lysed on ice for 30 min with 100 µL of protein lysis buffer. After centrifugation at 12,000 rpm for 10 min, the supernatant was collected. A total of 200 µL of the supernatant was reserved as input. The remaining 1 mL of lysate was incubated overnight at 4 °C with 2 µL of VP6 monoclonal antibody. Then, 40 µL of protein A/G agarose beads were added, and the mixture was incubated at 4 °C for an additional 6 h to allow antibody-antigen complexes to bind to the beads. After centrifugation at 2,500 rpm for 5 min, the pellet was collected and washed 5 times with PBS. Finally, loading buffer was added and boiled in a metal bath at 100 °C for 10 min. The input and immunoprecipitated samples were then analyzed by Western blotting.
Determination of RNA decay time
After 48 h of transfection, the culture medium was removed, and complete culture medium containing 5 µg/mL actinomycin D was added. The cells were then incubated in an incubator at 37℃ and 5% CO2 for 0, 3, and 6 h, respectively. Following this, total cellular RNA was extracted and reverse-transcribed into cDNA. The mRNA levels of transcripts were detected by qPCR. The rate of mRNA degradation (K) was calculated using the formula: log2 (At/A0) = -Kt [2], where t is the transcription inhibition time, At and A0 represent the mRNA levels at time t h and 0 h respectively, and k is determined by comparing the mRNA levels at 3 and 6 h with those at 0 h. Half-time of mRNA (T1/2) was calculated as follows: T1/2 = 2ln2/(k3 h + k6 h).
RIP-qPCR
The RIP-qPCR assay was conducted with modifications based on a previously described protocol [19]. Cells were lysed in lysis buffer containing 1% SUPERaseln, 1% protease inhibitor cocktail, 150 mM KCl, 0.5 mM DTT, 2 mM EDTA, 10 mM Tris-HCl, and 0.5% NP-40. Subsequently, immunocomplexes were formed by incubating 5 µg of YTHDF2 antibody or rabbit IgG with 40 µL of protein A magnetic beads for 1 h at 4 °C. The cell lysate was then co-incubated with protein A-anti YTHDF2 or protein A-IgG complexes for 3 h at 4 °C. The mixture was washed with pre-chilled NT2 buffer (200 mM NaCl, 0.1% SUPERaseln, 0.1% protease inhibitor cocktail, 50 mM Tris-HCl, 2 mM EDTA, 0.05% NP-40, 0.5 mM DTT). RNA was then eluted through proteinase K treatment at 55℃ for 40 min. Finally, RNA was isolated using Trizol reagent, and qRT-PCR was performed using SYBR fluorescent dye system.
Statistical analysis
The results were expressed as mean ± standard error of the mean (SEM). SPSS 20 software was used to perform independent sample t-test between two groups. GraphPad Prism 8.0 software was utilized for data visualization. Statistical significance was denoted by * for P < 0.05, ** for P < 0.01, *** for P < 0.001 between groups. Post hoc differences among treatment groups were assessed using Duncan’s multiple-range test.

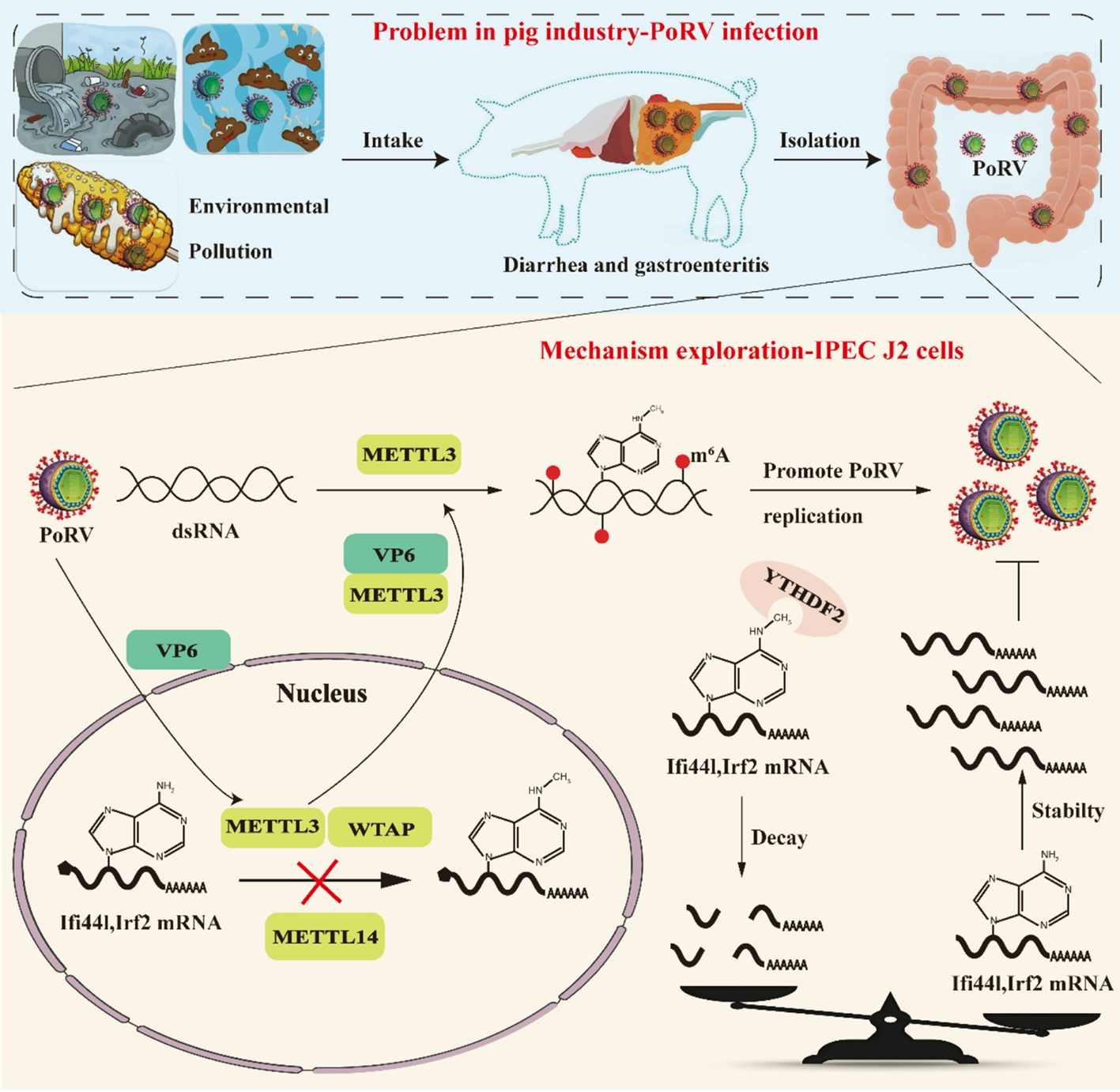


Comments (0)